

Regulation of cellular communication by signaling microdomains in the blood vessel wall
- PMID: 24671377
- PMCID: PMC3973613
- DOI: 10.1124/pr.112.007351
Regulation of cellular communication by signaling microdomains in the blood vessel wall
Abstract
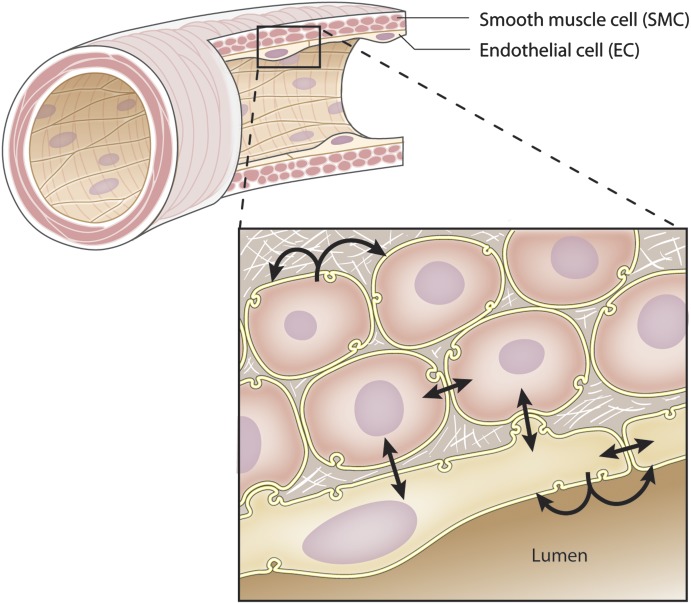
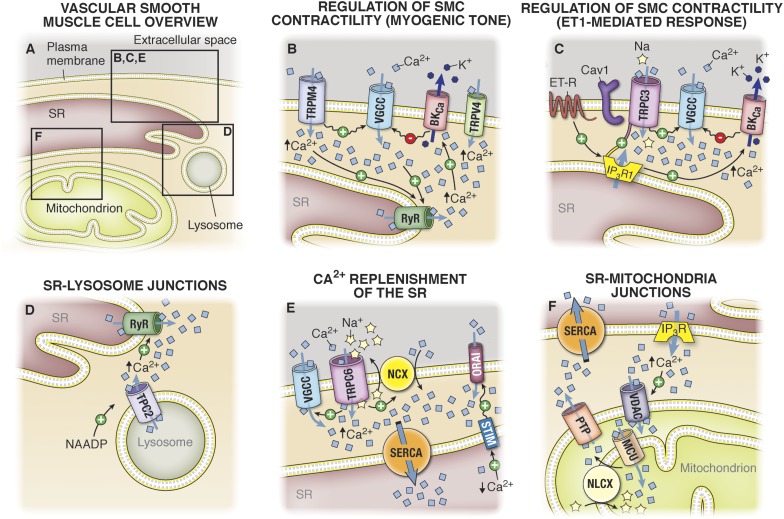
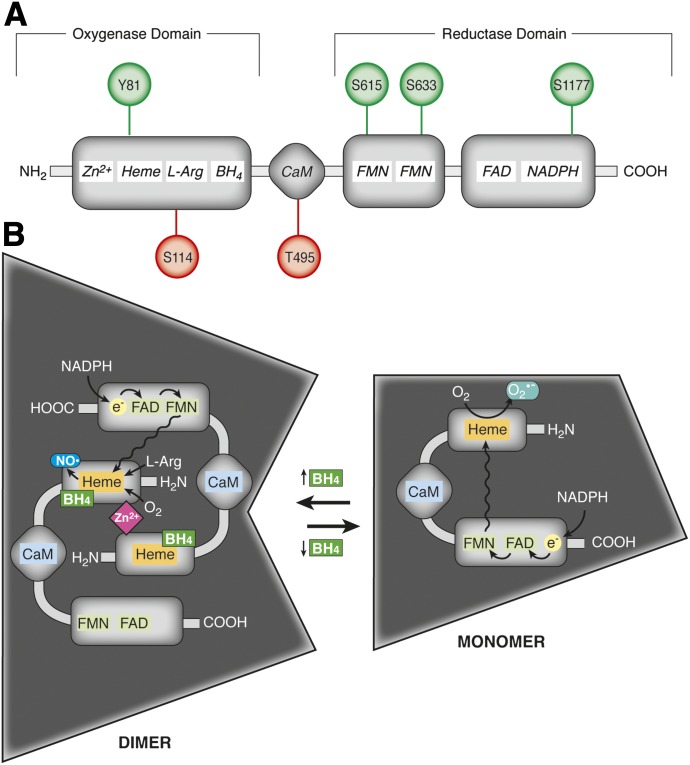
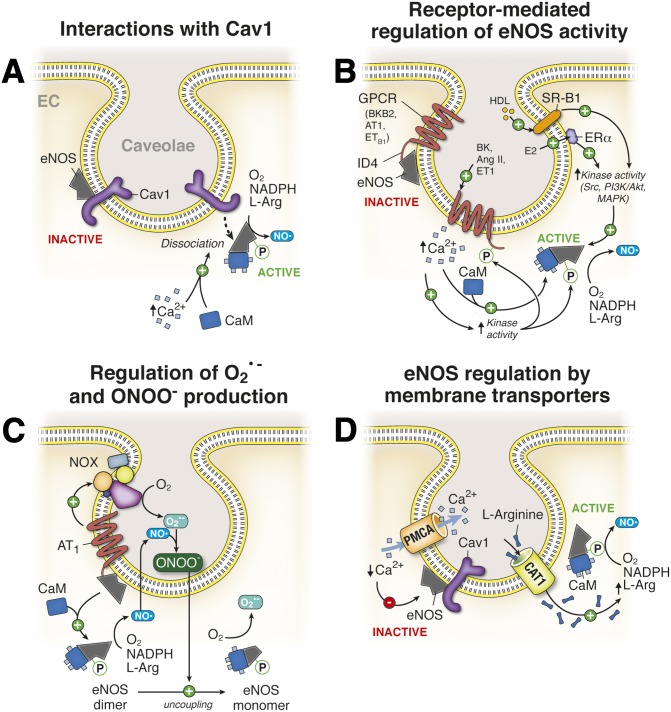
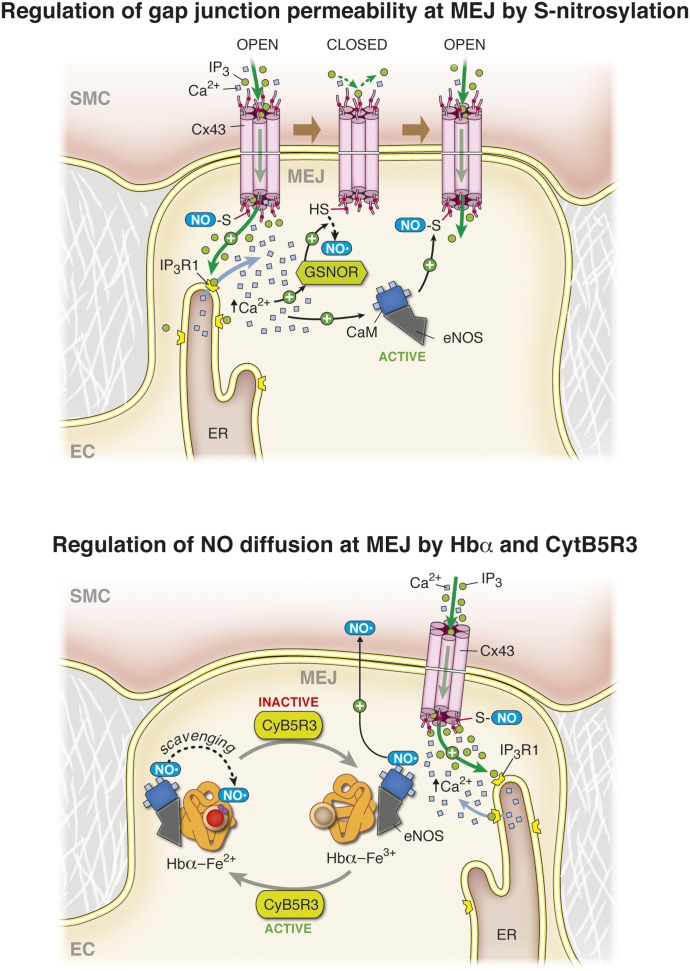
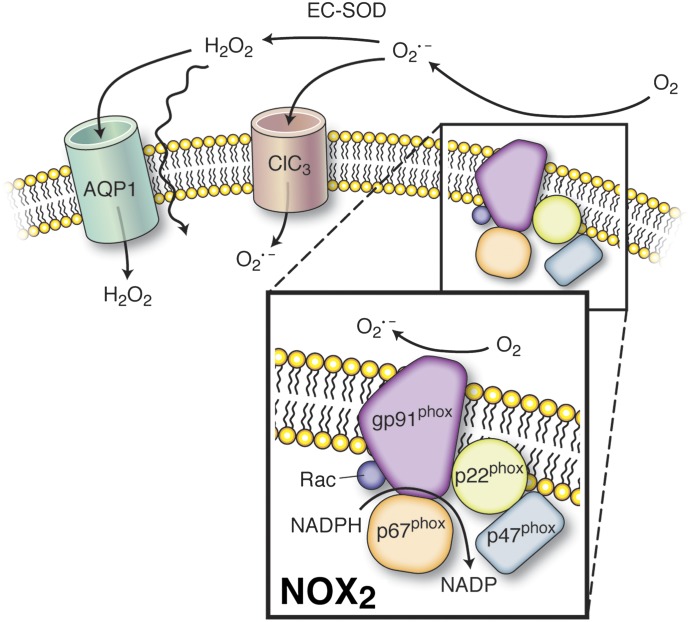
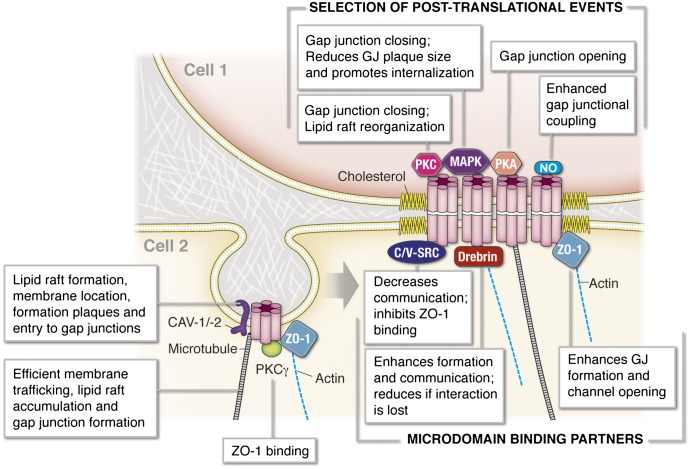
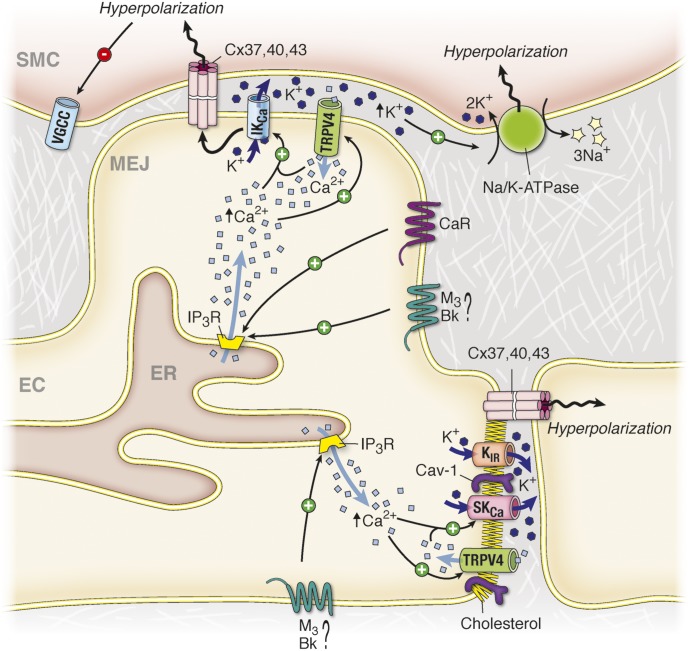
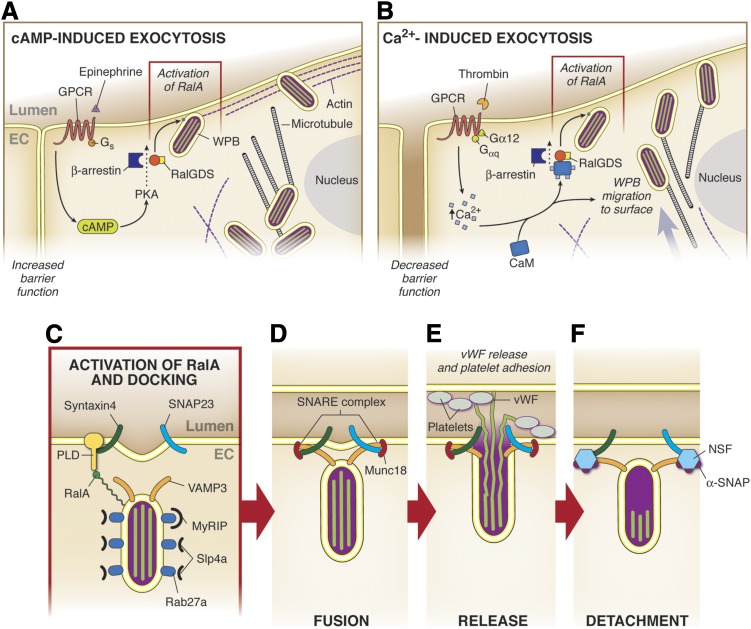
It has become increasingly clear that the accumulation of proteins in specific regions of the plasma membrane can facilitate cellular communication. These regions, termed signaling microdomains, are found throughout the blood vessel wall where cellular communication, both within and between cell types, must be tightly regulated to maintain proper vascular function. We will define a cellular signaling microdomain and apply this definition to the plethora of means by which cellular communication has been hypothesized to occur in the blood vessel wall. To that end, we make a case for three broad areas of cellular communication where signaling microdomains could play an important role: 1) paracrine release of free radicals and gaseous molecules such as nitric oxide and reactive oxygen species; 2) role of ion channels including gap junctions and potassium channels, especially those associated with the endothelium-derived hyperpolarization mediated signaling, and lastly, 3) mechanism of exocytosis that has considerable oversight by signaling microdomains, especially those associated with the release of von Willebrand factor. When summed, we believe that it is clear that the organization and regulation of signaling microdomains is an essential component to vessel wall function.
Figures
References
-
- Abo A, Pick E, Hall A, Totty N, Teahan CG, Segal AW. (1991) Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 353:668–670 - PubMed
-
- Adeagbo AS, Triggle CR. (1993) Varying extracellular [K+]: a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J Cardiovasc Pharmacol 21:423–429 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
