

Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression
- PMID: 24671853
- PMCID: PMC4032861
- DOI: 10.1101/gr.169417.113
Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression
Abstract
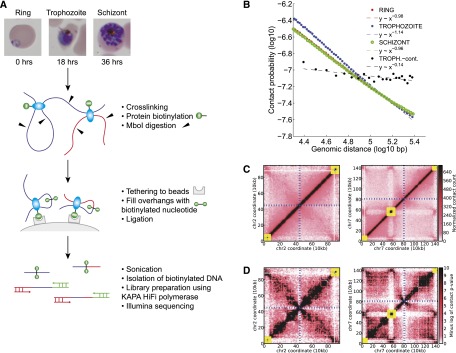
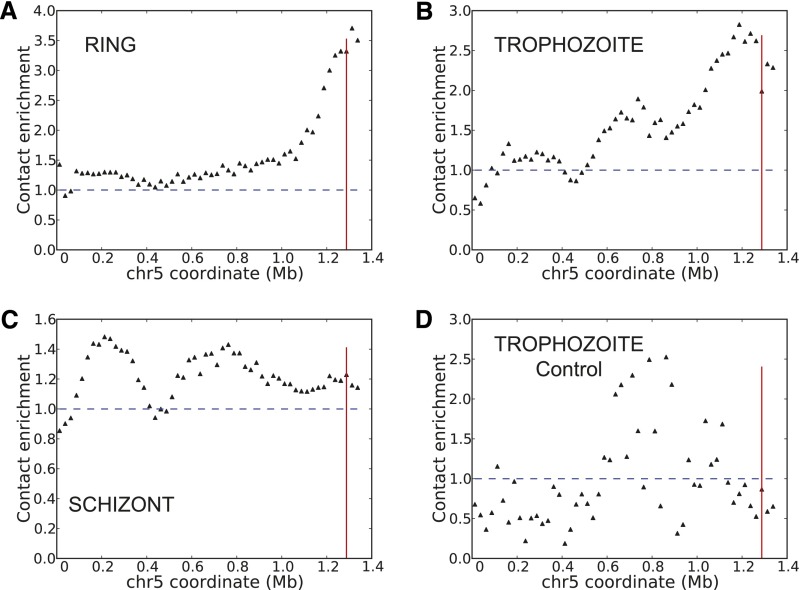
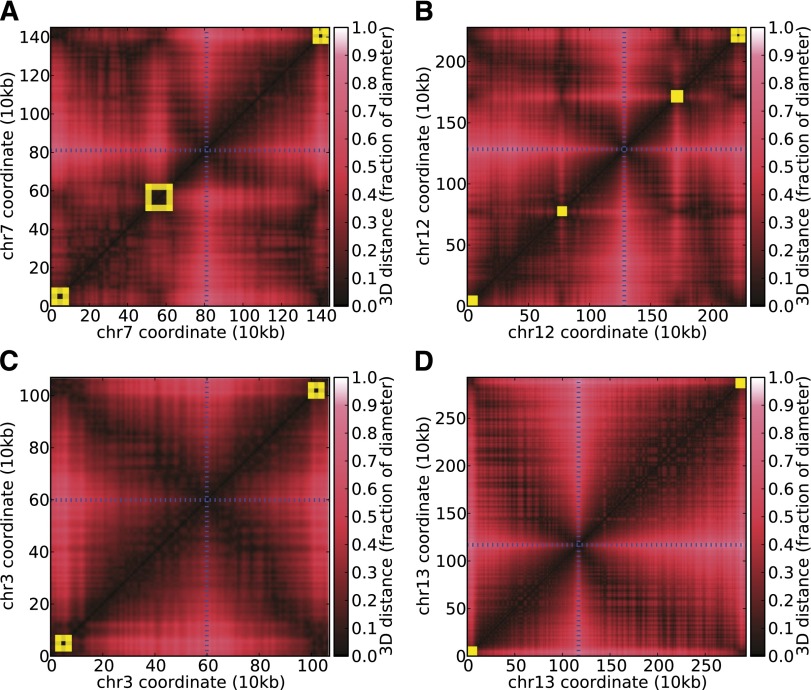
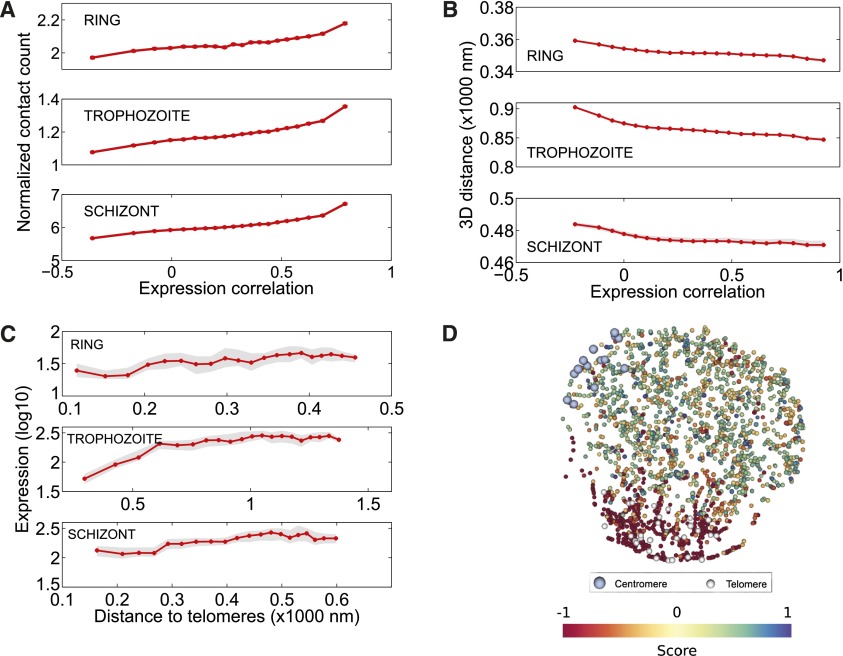
The development of the human malaria parasite Plasmodium falciparum is controlled by coordinated changes in gene expression throughout its complex life cycle, but the corresponding regulatory mechanisms are incompletely understood. To study the relationship between genome architecture and gene regulation in Plasmodium, we assayed the genome architecture of P. falciparum at three time points during its erythrocytic (asexual) cycle. Using chromosome conformation capture coupled with next-generation sequencing technology (Hi-C), we obtained high-resolution chromosomal contact maps, which we then used to construct a consensus three-dimensional genome structure for each time point. We observed strong clustering of centromeres, telomeres, ribosomal DNA, and virulence genes, resulting in a complex architecture that cannot be explained by a simple volume exclusion model. Internal virulence gene clusters exhibit domain-like structures in contact maps, suggesting that they play an important role in the genome architecture. Midway during the erythrocytic cycle, at the highly transcriptionally active trophozoite stage, the genome adopts a more open chromatin structure with increased chromosomal intermingling. In addition, we observed reduced expression of genes located in spatial proximity to the repressive subtelomeric center, and colocalization of distinct groups of parasite-specific genes with coordinated expression profiles. Overall, our results are indicative of a strong association between the P. falciparum spatial genome organization and gene expression. Understanding the molecular processes involved in genome conformation dynamics could contribute to the discovery of novel antimalarial strategies.
© 2014 Ay et al.; Published by Cold Spring Harbor Laboratory Press.
Figures


References
-
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
-
- Bach FR, Jordan MI 2002. Kernel independent component analysis. J Mach Learn Res 3: 1–48
-
- Bannister LH, Margos G, Hopkins JM 2005. Making a home for Plasmodium post-genomics: ultrastructural organization of the blood stages. In Molecular approaches to malaria, pp. 24–49. ASM Press, Washington, DC
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases