

NEMO is essential for Kaposi's sarcoma-associated herpesvirus-encoded vFLIP K13-induced gene expression and protection against death receptor-induced cell death, and its N-terminal 251 residues are sufficient for this process
- PMID: 24672029
- PMCID: PMC4093867
- DOI: 10.1128/JVI.00028-14
NEMO is essential for Kaposi's sarcoma-associated herpesvirus-encoded vFLIP K13-induced gene expression and protection against death receptor-induced cell death, and its N-terminal 251 residues are sufficient for this process
Abstract
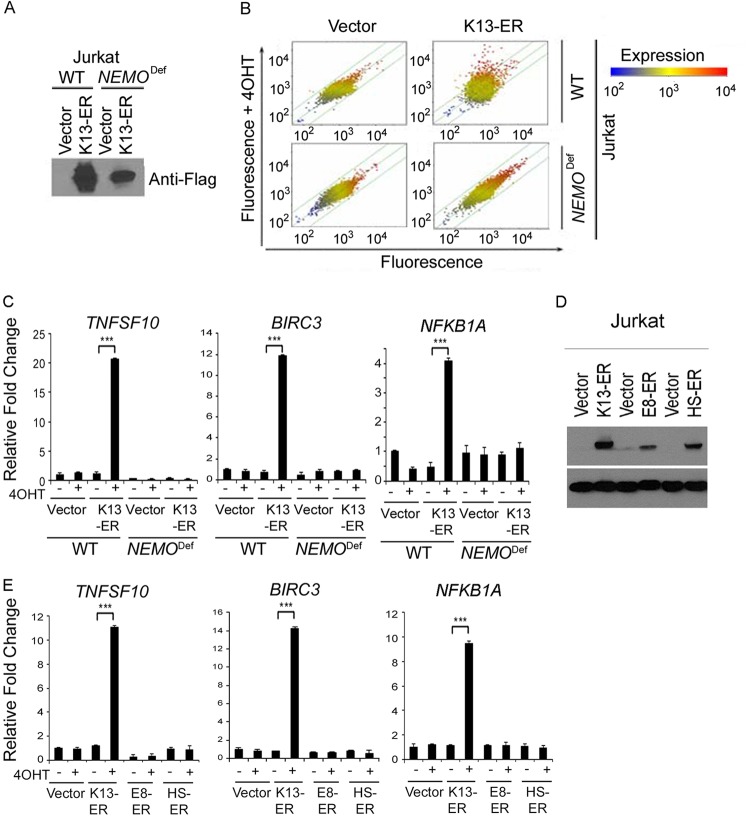
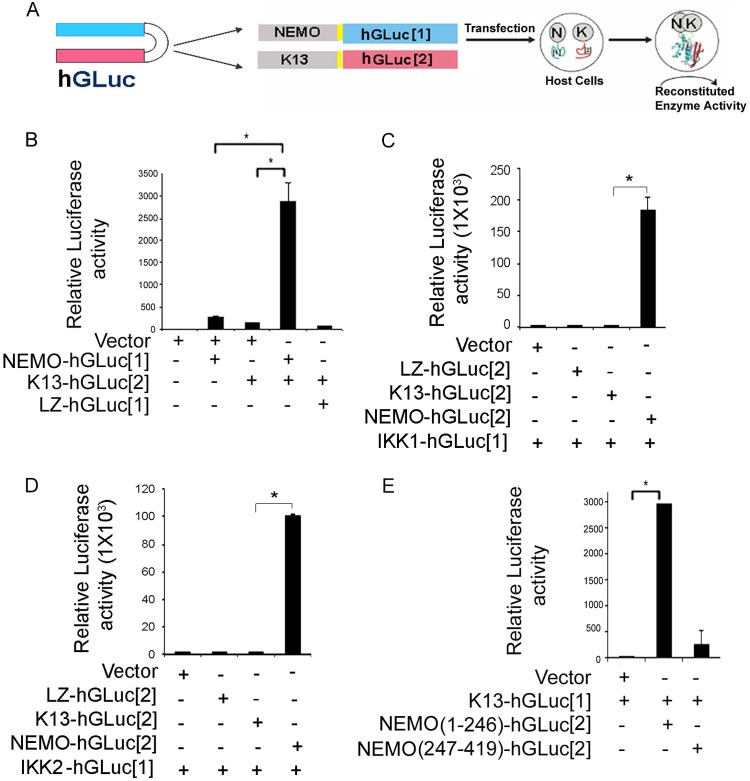
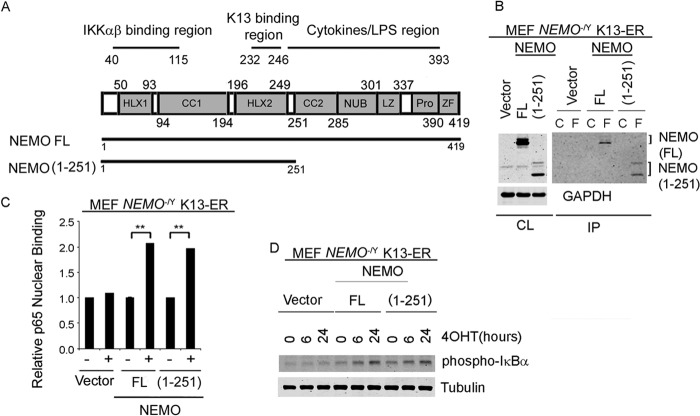
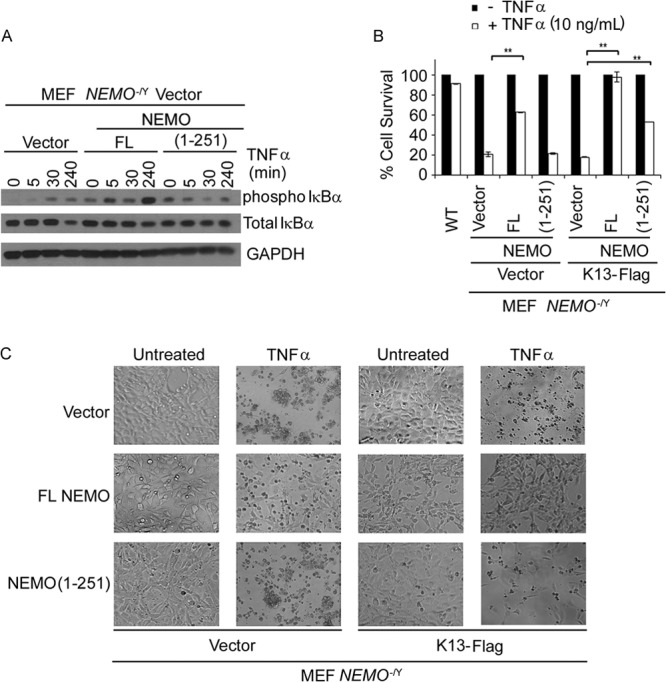
Kaposi's sarcoma-associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 was originally believed to protect virally infected cells against death receptor-induced apoptosis by interfering with caspase 8/FLICE activation. Subsequent studies revealed that K13 also activates the NF-κB pathway by binding to the NEMO/inhibitor of NF-κB (IκB) kinase gamma (IKKγ) subunit of an IKK complex and uses this pathway to modulate the expression of genes involved in cellular survival, proliferation, and the inflammatory response. However, it is not clear if K13 can also induce gene expression independently of NEMO/IKKγ. The minimum region of NEMO that is sufficient for supporting K13-induced NF-κB has not been delineated. Furthermore, the contribution of NEMO and NF-κB to the protective effect of K13 against death receptor-induced apoptosis remains to be determined. In this study, we used microarray analysis on K13-expressing wild-type and NEMO-deficient cells to demonstrate that NEMO is required for modulation of K13-induced genes. Reconstitution of NEMO-null cells revealed that the N-terminal 251 amino acid residues of NEMO are sufficient for supporting K13-induced NF-κB but fail to support tumor necrosis factor alpha (TNF-α)-induced NF-κB. K13 failed to protect NEMO-null cells against TNF-α-induced cell death but protected those reconstituted with the NEMO mutant truncated to include only the N-terminal 251 amino acid residues [the NEMO(1-251) mutant]. Taken collectively, our results demonstrate that NEMO is required for modulation of K13-induced genes and the N-terminal 251 amino acids of NEMO are sufficient for supporting K13-induced NF-κB. Finally, the ability of K13 to protect against TNF-α-induced cell death is critically dependent on its ability to interact with NEMO and activate NF-κB.
Importance: Kaposi's sarcoma-associated herpesvirus-encoded vFLIP K13 is believed to protect virally infected cells against death receptor-induced apoptosis and to activate the NF-κB pathway by binding to adaptor protein NEMO/IKKγ. However, whether K13 can also induce gene expression independently of NEMO and the minimum region of NEMO that is sufficient for supporting K13-induced NF-κB remain to be delineated. Furthermore, the contribution of NEMO and NF-κB to the protective effect of K13 against death receptor-induced apoptosis is not clear. We demonstrate that NEMO is required for modulation of K13-induced genes and its N-terminal 251 amino acids are sufficient for supporting K13-induced NF-κB. The ability of K13 to protect against TNF-α-induced cell death is critically dependent on its ability to interact with NEMO and activate NF-κB. Our results suggest that K13-based gene therapy approaches may have utility for the treatment of patients with NEMO mutations and immunodeficiency.
Figures
Similar articles
-
Kaposi's sarcoma associated herpesvirus encoded viral FLICE inhibitory protein K13 activates NF-κB pathway independent of TRAF6, TAK1 and LUBAC.PLoS One. 2012;7(5):e36601. doi: 10.1371/journal.pone.0036601. Epub 2012 May 8. PLoS One. 2012. PMID: 22590573 Free PMC article.
-
Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein-induced NF-kappaB activation.J Biol Chem. 2003 Dec 26;278(52):52406-11. doi: 10.1074/jbc.M307308200. Epub 2003 Oct 15. J Biol Chem. 2003. PMID: 14561765
-
Novel Phosphorylations of IKKγ/NEMO.mBio. 2012 Nov 6;3(6):e00411-12. doi: 10.1128/mBio.00411-12. mBio. 2012. PMID: 23131831 Free PMC article.
-
Kaposi's sarcoma associated herpes virus-encoded viral FLICE inhibitory protein activates transcription from HIV-1 Long Terminal Repeat via the classical NF-kappaB pathway and functionally cooperates with Tat.Retrovirology. 2005 Feb 15;2:9. doi: 10.1186/1742-4690-2-9. Retrovirology. 2005. PMID: 15713234 Free PMC article.
-
The zinc finger domain of IKKγ (NEMO) protein in health and disease.J Cell Mol Med. 2010 Oct;14(10):2404-14. doi: 10.1111/j.1582-4934.2010.01054.x. J Cell Mol Med. 2010. PMID: 20345847 Free PMC article. Review.
Cited by
-
Narciclasine, an isocarbostyril alkaloid, has preferential activity against primary effusion lymphoma.Sci Rep. 2020 Mar 31;10(1):5712. doi: 10.1038/s41598-020-62690-9. Sci Rep. 2020. PMID: 32235878 Free PMC article.
-
CRISPR screens identify novel regulators of cFLIP dependency and ligand-independent, TRAIL-R1-mediated cell death.Cell Death Differ. 2023 May;30(5):1221-1234. doi: 10.1038/s41418-023-01133-0. Epub 2023 Feb 18. Cell Death Differ. 2023. PMID: 36801923 Free PMC article.
-
Azidothymidine Sensitizes Primary Effusion Lymphoma Cells to Kaposi Sarcoma-Associated Herpesvirus-Specific CD4+ T Cell Control and Inhibits vIRF3 Function.PLoS Pathog. 2016 Nov 28;12(11):e1006042. doi: 10.1371/journal.ppat.1006042. eCollection 2016 Nov. PLoS Pathog. 2016. PMID: 27893813 Free PMC article.
-
A novel thermostable beetle luciferase based cytotoxicity assay.Sci Rep. 2021 May 11;11(1):10002. doi: 10.1038/s41598-021-89404-z. Sci Rep. 2021. PMID: 33976304 Free PMC article.
-
Kaposi's Sarcoma-Associated Herpesvirus, the Etiological Agent of All Epidemiological Forms of Kaposi's Sarcoma.Cancers (Basel). 2021 Dec 9;13(24):6208. doi: 10.3390/cancers13246208. Cancers (Basel). 2021. PMID: 34944828 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
