

The Thr205 phosphorylation site within respiratory syncytial virus matrix (M) protein modulates M oligomerization and virus production
- PMID: 24672034
- PMCID: PMC4093874
- DOI: 10.1128/JVI.03856-13
The Thr205 phosphorylation site within respiratory syncytial virus matrix (M) protein modulates M oligomerization and virus production
Abstract
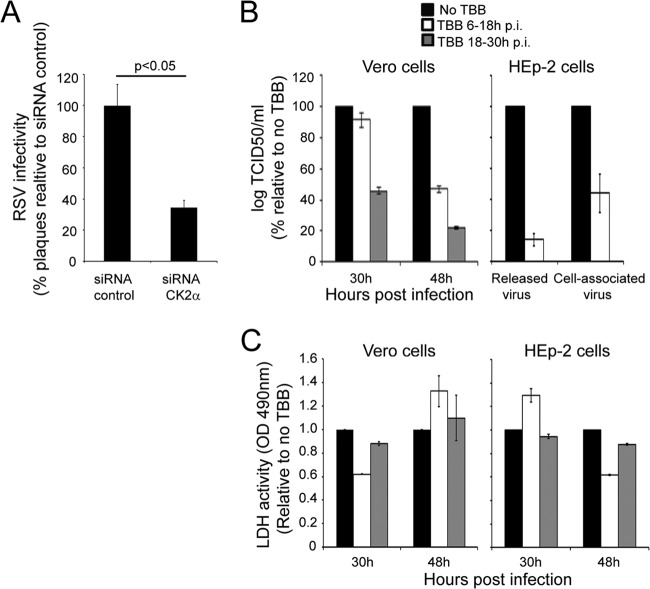
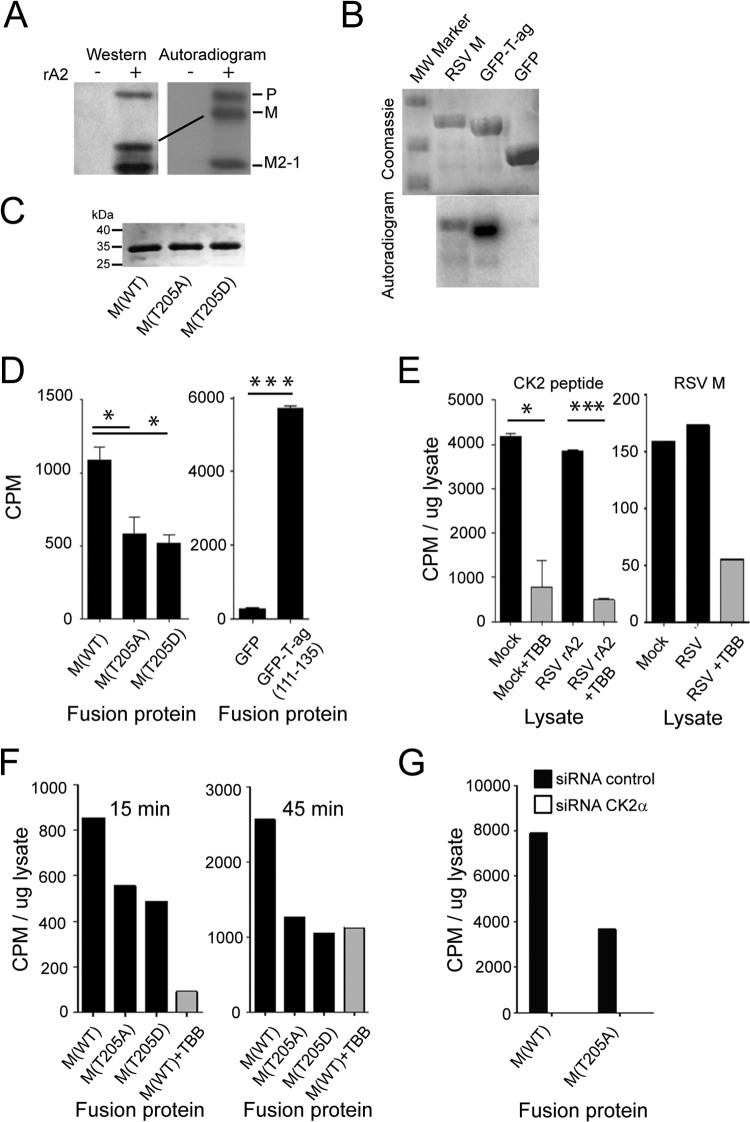
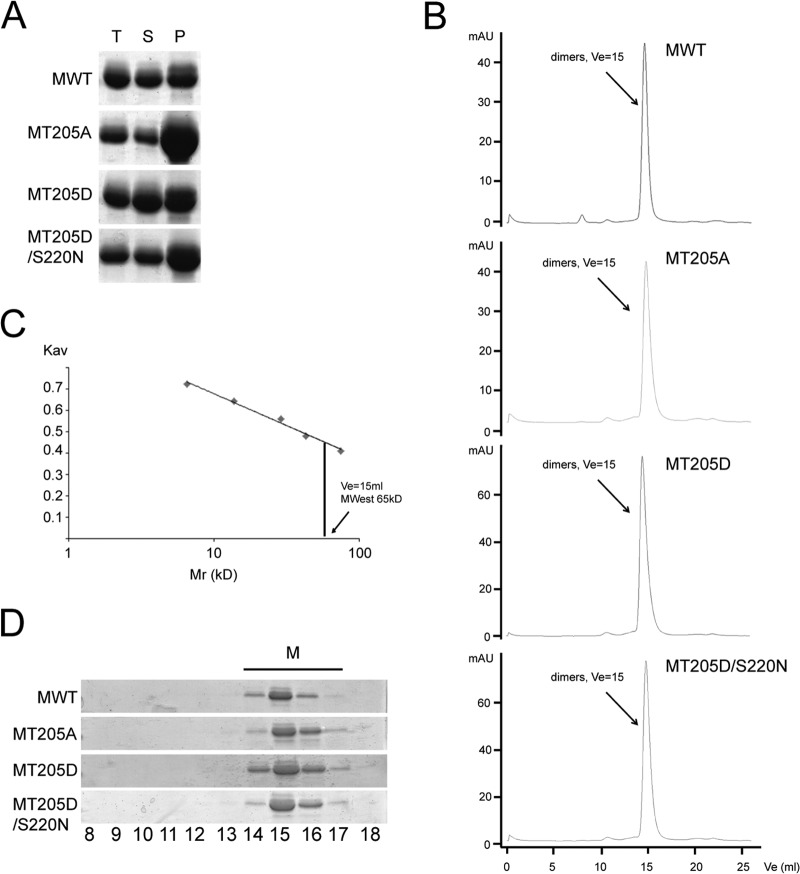
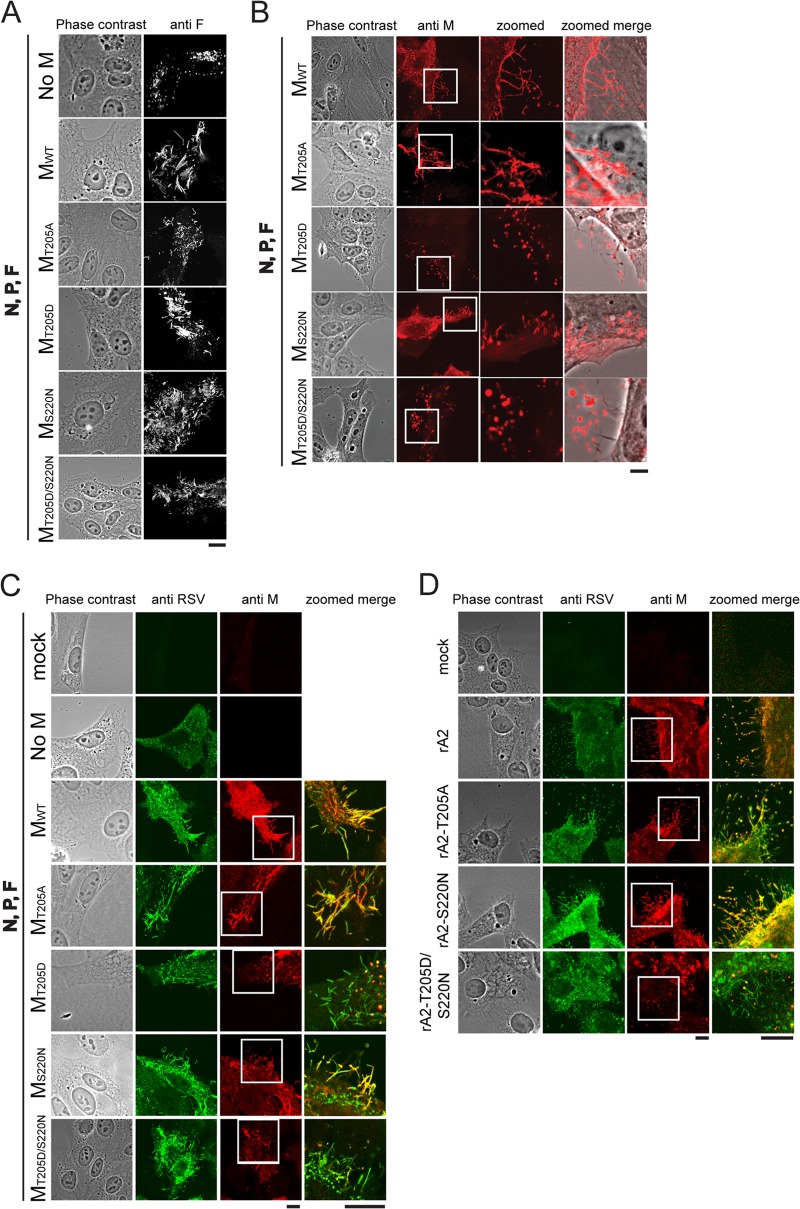
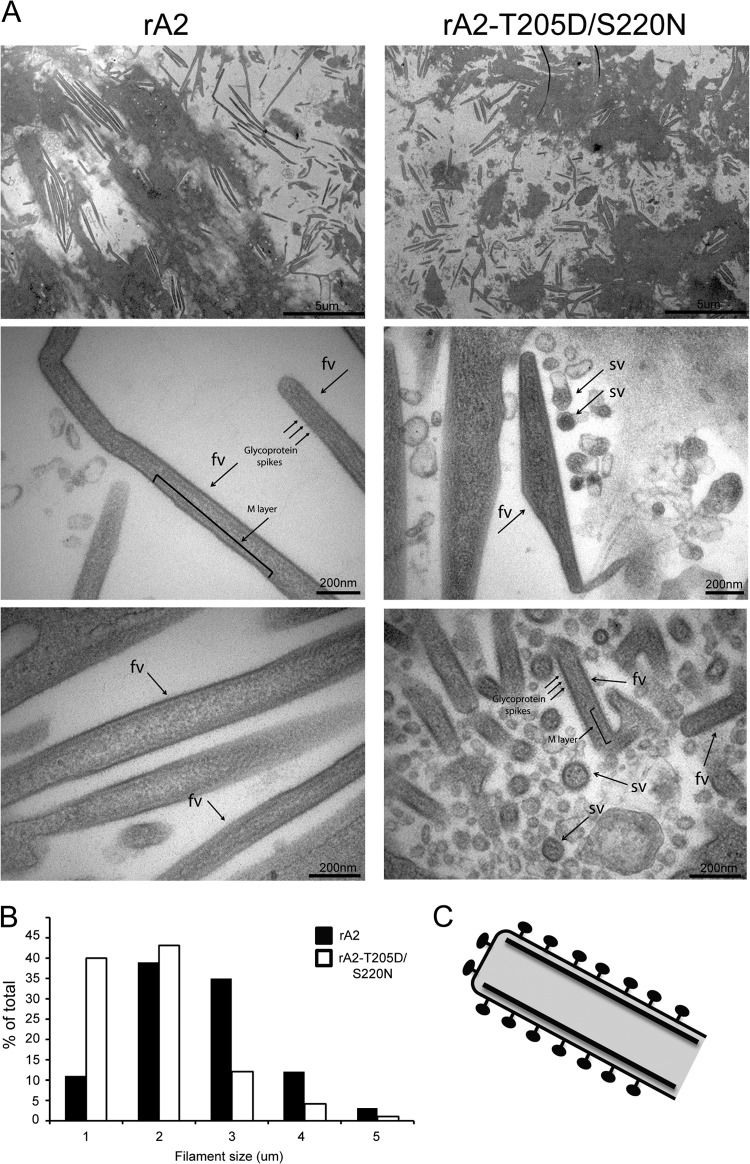
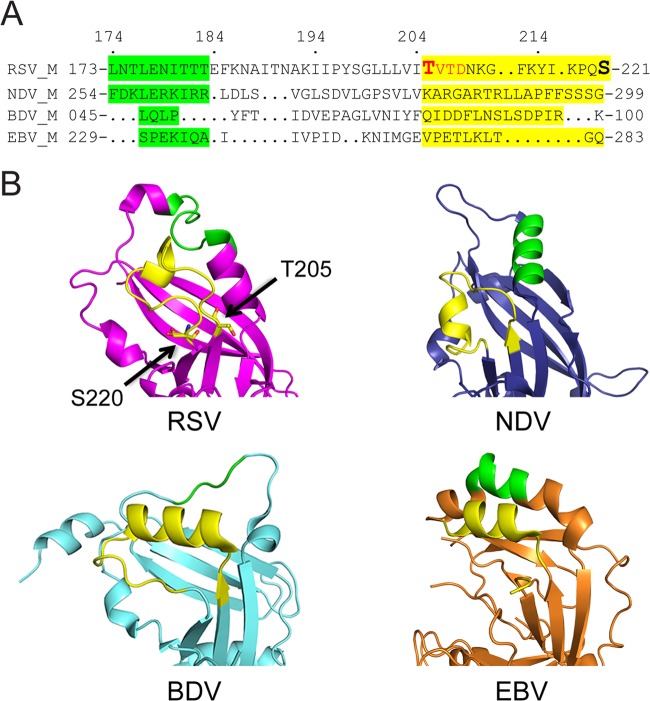
Human respiratory syncytial virus (RSV) is the most common cause of bronchiolitis and pneumonia in infants and the elderly worldwide; however, there is no licensed RSV vaccine or effective drug treatment available. The RSV matrix (M) protein plays key roles in virus assembly and budding, but the protein interactions that govern budding of infectious virus are not known. In this study, we focus on M protein and identify a key phosphorylation site (Thr205) in M that is critical for RSV infectious virus production. Recombinant virus with a nonphosphorylatable alanine (Ala) residue at the site was markedly attenuated, whereas virus with a phosphomimetic aspartate (Asp) resulted in a nonviable virus which could only be recovered with an additional mutation in M (serine to asparagine at position 220), strongly implying that Thr205 is critical for viral infectivity. Experiments in vitro showed that mutation of Thr205 does not affect M stability or the ability to form dimers but implicate an effect on higher-order oligomer assembly. In transfected and infected cells, Asp substitution of Thr205 appeared to impair M oligomerization; typical filamentous structures still formed at the plasma membrane, but M assembly during the ensuing elongation process seemed to be impaired, resulting in shorter and more branched filaments as observed using electron microscopy (EM). Our data thus imply for the first time that M oligomerization, regulated by a negative charge at Thr205, may be critical to production of infectious RSV.
Importance: We show here for the first time that RSV M's role in virus assembly/release is strongly dependent on threonine 205 (Thr205), a consensus site for CK2, which appears to play a key regulatory role in modulating M oligomerization and association with virus filaments. Our analysis indicates that T205 mutations do not impair M dimerization or viruslike filament formation per se but rather the ability of M to assemble in ordered fashion on the viral filaments themselves. This appears to impact in turn upon the infectivity of released virus rather than on virus production or release itself. Thus, M oligomerization would appear to be a target of interest for the development of anti-RSV agents; further, the recombinant T205-substituted mutant viruses described here would appear to be the first RSV mutants affected in viral maturation to our knowledge and hence of considerable interest for vaccine approaches in the future.
Figures
Similar articles
-
Dimerization of matrix protein is required for budding of respiratory syncytial virus.J Virol. 2015 Apr;89(8):4624-35. doi: 10.1128/JVI.03500-14. Epub 2015 Feb 11. J Virol. 2015. PMID: 25673702 Free PMC article.
-
A critical phenylalanine residue in the respiratory syncytial virus fusion protein cytoplasmic tail mediates assembly of internal viral proteins into viral filaments and particles.mBio. 2012 Feb 7;3(1):e00270-11. doi: 10.1128/mBio.00270-11. Print 2012. mBio. 2012. PMID: 22318318 Free PMC article.
-
Respiratory Syncytial Virus Matrix Protein-Chromatin Association Is Key to Transcriptional Inhibition in Infected Cells.Cells. 2021 Oct 18;10(10):2786. doi: 10.3390/cells10102786. Cells. 2021. PMID: 34685766 Free PMC article.
-
Evaluation of the Safety and Immune Efficacy of Recombinant Human Respiratory Syncytial Virus Strain Long Live Attenuated Vaccine Candidates.Virol Sin. 2021 Aug;36(4):706-720. doi: 10.1007/s12250-021-00345-3. Epub 2021 Feb 9. Virol Sin. 2021. PMID: 33559831 Free PMC article. Review.
-
Host cytoskeleton in respiratory syncytial virus assembly and budding.Virol J. 2016 Sep 26;13(1):161. doi: 10.1186/s12985-016-0618-z. Virol J. 2016. PMID: 27670781 Free PMC article. Review.
Cited by
-
The actin-binding protein palladin associates with the respiratory syncytial virus matrix protein.J Virol. 2024 Oct 22;98(10):e0143524. doi: 10.1128/jvi.01435-24. Epub 2024 Oct 3. J Virol. 2024. PMID: 39360826 Free PMC article.
-
The Morphology and Assembly of Respiratory Syncytial Virus Revealed by Cryo-Electron Tomography.Viruses. 2018 Aug 20;10(8):446. doi: 10.3390/v10080446. Viruses. 2018. PMID: 30127286 Free PMC article.
-
Expression and Purification of Recombinant Respiratory Syncytial Virus Proteins.Methods Mol Biol. 2025;2948:137-157. doi: 10.1007/978-1-0716-4666-3_10. Methods Mol Biol. 2025. PMID: 40879907
-
New host factors important for respiratory syncytial virus (RSV) replication revealed by a novel microfluidics screen for interactors of matrix (M) protein.Mol Cell Proteomics. 2015 Mar;14(3):532-43. doi: 10.1074/mcp.M114.044107. Epub 2015 Jan 2. Mol Cell Proteomics. 2015. PMID: 25556234 Free PMC article.
-
Interaction of Human Respiratory Syncytial Virus (HRSV) Matrix Protein with Resveratrol Shows Antiviral Effect.Int J Mol Sci. 2024 Nov 28;25(23):12790. doi: 10.3390/ijms252312790. Int J Mol Sci. 2024. PMID: 39684498 Free PMC article.
References
-
- Collins PL, Crowe JE. 2007. Respiratory syncytial virus and human metapneumovirus, p 1601–1646 In Knipe H, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott, Williams, and Wilkins, Baltimore, MD
-
- Garcia J, Garcia-Barreno B, Vivo A, Melero JA. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195:243–247. 10.1006/viro.1993.1366 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
