

TSSAR: TSS annotation regime for dRNA-seq data
- PMID: 24674136
- PMCID: PMC4098767
- DOI: 10.1186/1471-2105-15-89
TSSAR: TSS annotation regime for dRNA-seq data
Abstract
Background: Differential RNA sequencing (dRNA-seq) is a high-throughput screening technique designed to examine the architecture of bacterial operons in general and the precise position of transcription start sites (TSS) in particular. Hitherto, dRNA-seq data were analyzed by visualizing the sequencing reads mapped to the reference genome and manually annotating reliable positions. This is very labor intensive and, due to the subjectivity, biased.
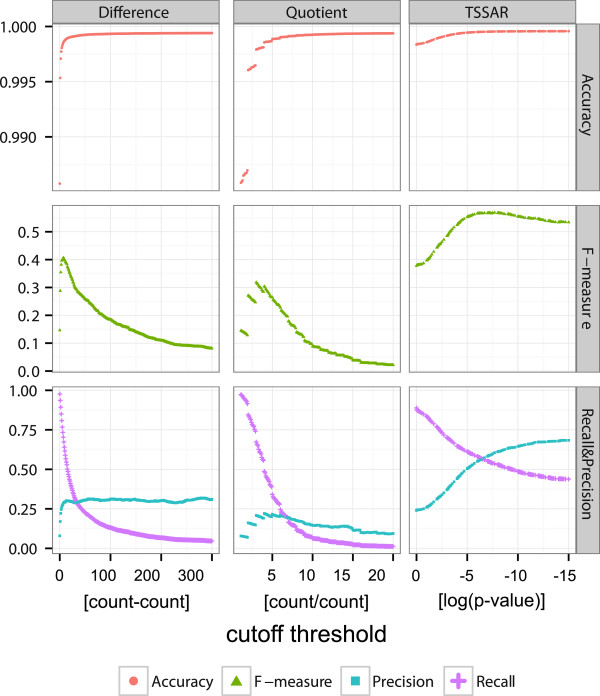
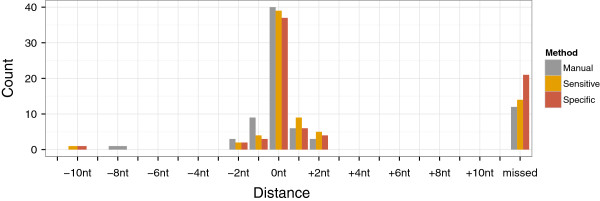
Results: Here, we present TSSAR, a tool for automated de novo TSS annotation from dRNA-seq data that respects the statistics of dRNA-seq libraries. TSSAR uses the premise that the number of sequencing reads starting at a certain genomic position within a transcriptional active region follows a Poisson distribution with a parameter that depends on the local strength of expression. The differences of two dRNA-seq library counts thus follow a Skellam distribution. This provides a statistical basis to identify significantly enriched primary transcripts.We assessed the performance by analyzing a publicly available dRNA-seq data set using TSSAR and two simple approaches that utilize user-defined score cutoffs. We evaluated the power of reproducing the manual TSS annotation. Furthermore, the same data set was used to reproduce 74 experimentally validated TSS in H. pylori from reliable techniques such as RACE or primer extension. Both analyses showed that TSSAR outperforms the static cutoff-dependent approaches.
Conclusions: Having an automated and efficient tool for analyzing dRNA-seq data facilitates the use of the dRNA-seq technique and promotes its application to more sophisticated analysis. For instance, monitoring the plasticity and dynamics of the transcriptomal architecture triggered by different stimuli and growth conditions becomes possible.The main asset of a novel tool for dRNA-seq analysis that reaches out to a broad user community is usability. As such, we provide TSSAR both as intuitive RESTful Web service ( http://rna.tbi.univie.ac.at/TSSAR) together with a set of post-processing and analysis tools, as well as a stand-alone version for use in high-throughput dRNA-seq data analysis pipelines.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
