

Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolon-intestinal hypoperistalsis syndrome
- PMID: 24676022
- PMCID: PMC3967950
- DOI: 10.1371/journal.pgen.1004258
Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolon-intestinal hypoperistalsis syndrome
Abstract
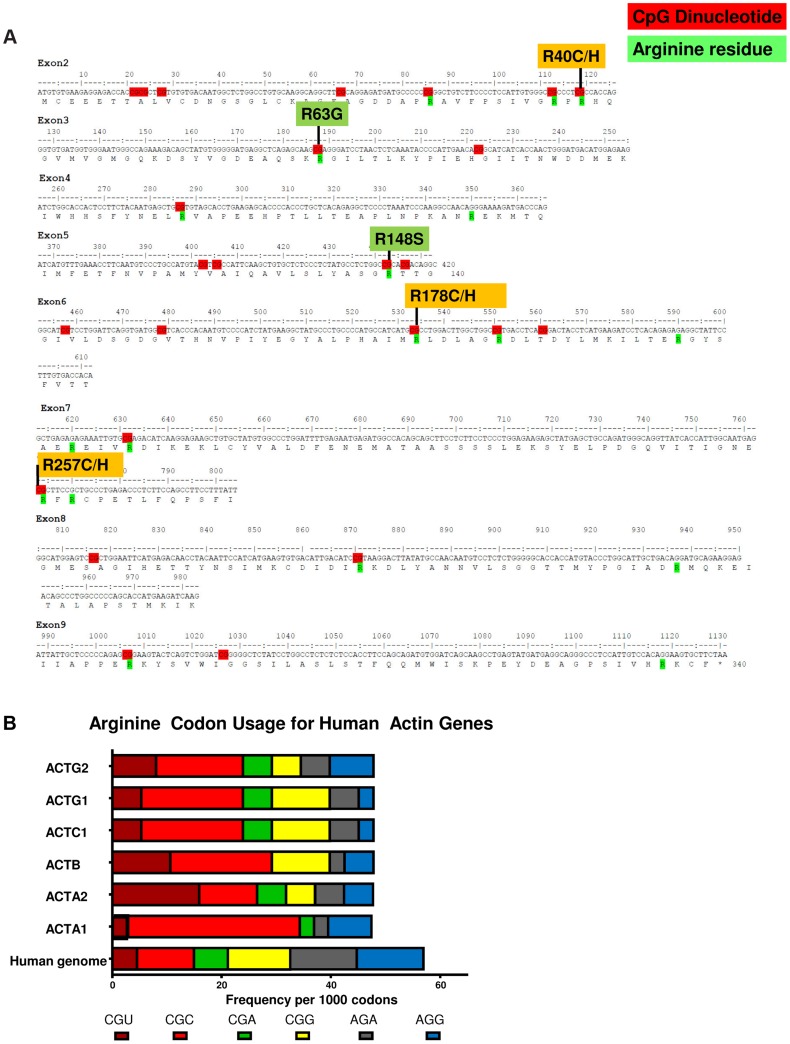
Megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS) is a rare disorder of enteric smooth muscle function affecting the intestine and bladder. Patients with this severe phenotype are dependent on total parenteral nutrition and urinary catheterization. The cause of this syndrome has remained a mystery since Berdon's initial description in 1976. No genes have been clearly linked to MMIHS. We used whole-exome sequencing for gene discovery followed by targeted Sanger sequencing in a cohort of patients with MMIHS and intestinal pseudo-obstruction. We identified heterozygous ACTG2 missense variants in 15 unrelated subjects, ten being apparent de novo mutations. Ten unique variants were detected, of which six affected CpG dinucleotides and resulted in missense mutations at arginine residues, perhaps related to biased usage of CpG containing codons within actin genes. We also found some of the same heterozygous mutations that we observed as apparent de novo mutations in MMIHS segregating in families with intestinal pseudo-obstruction, suggesting that ACTG2 is responsible for a spectrum of smooth muscle disease. ACTG2 encodes γ2 enteric actin and is the first gene to be clearly associated with MMIHS, suggesting an important role for contractile proteins in enteric smooth muscle disease.
Conflict of interest statement
The authors have read the journal's policy and have the following conflicts: The Department of Molecular and Human Genetics at Baylor College of Medicine (BCM) offers extensive genetic laboratory testing, and BCM derives revenue from this activity. The Department offers chromosomal microarray analysis, whole-exome sequencing, and many other tests.
Figures



References
-
- Berdon WE, Baker DH, Blanc WA, Gay B, Santulli TV, et al. (1976) Megacystis-microcolon-intestinal hypoperistalsis syndrome: a new cause of intestinal obstruction in the newborn. Report of radiologic findings in five newborn girls. AJR Am J Roentgenol 126: 957–964. - PubMed
-
- Gosemann JH, Puri P (2011) Megacystis microcolon intestinal hypoperistalsis syndrome: systematic review of outcome. Pediatr Surg Int 27: 1041–1046. - PubMed
-
- Kohler M, Pease PW, Upadhyay V (2004) Megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS) in siblings: case report and review of the literature. Eur J Pediatr Surg 14: 362–367. - PubMed
-
- Puri P, Shinkai M (2005) Megacystis microcolon intestinal hypoperistalsis syndrome. Semin Pediatr Surg 14: 58–63. - PubMed
-
- Schuler H, Nyakern M, Schutt CE, Lindberg U, Karlsson R (2000) Mutational analysis of arginine 177 in the nucleotide binding site of beta-actin. Eur J Biochem 267: 4054–4062. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
