

Coronary heart disease-associated variation in TCF21 disrupts a miR-224 binding site and miRNA-mediated regulation
- PMID: 24676100
- PMCID: PMC3967965
- DOI: 10.1371/journal.pgen.1004263
Coronary heart disease-associated variation in TCF21 disrupts a miR-224 binding site and miRNA-mediated regulation
Abstract
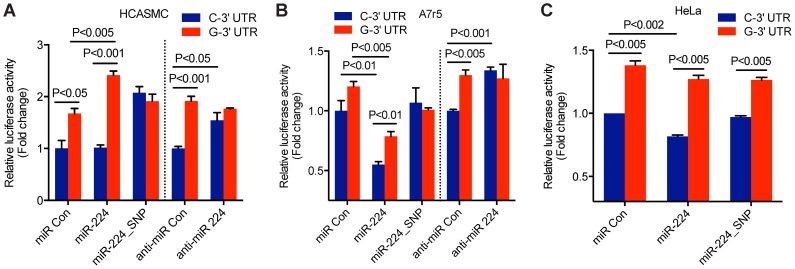
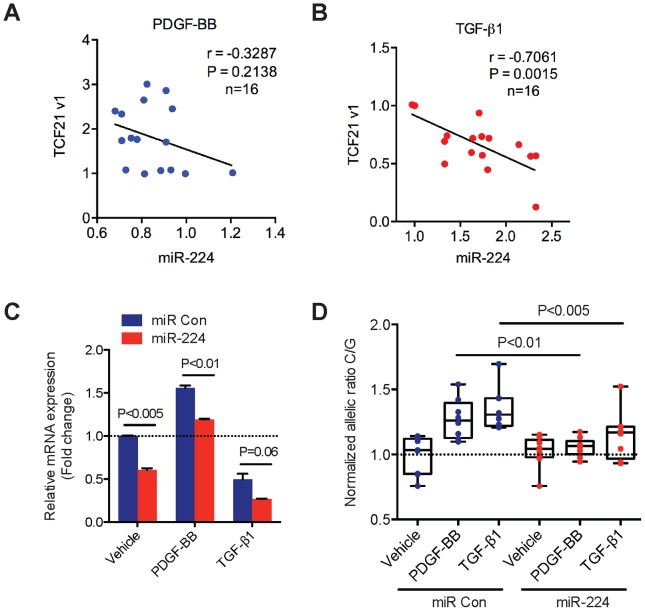
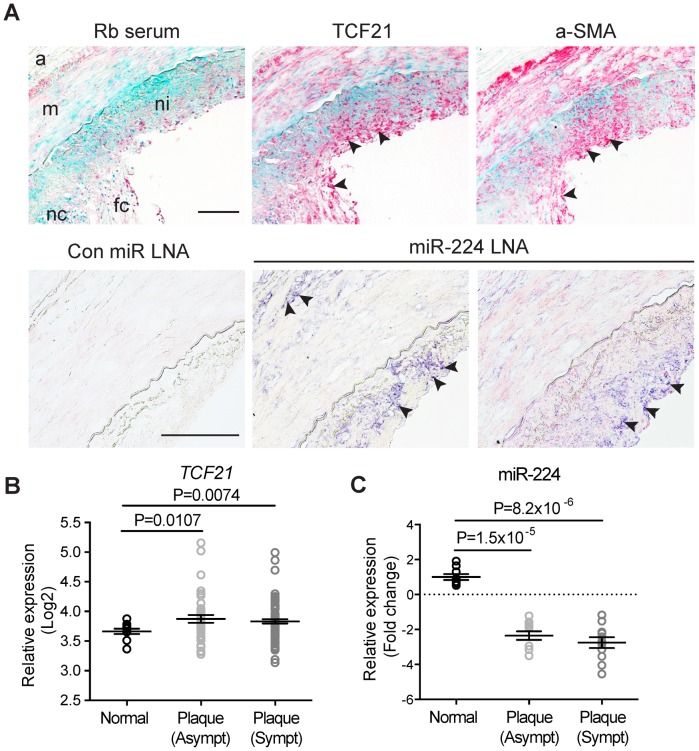
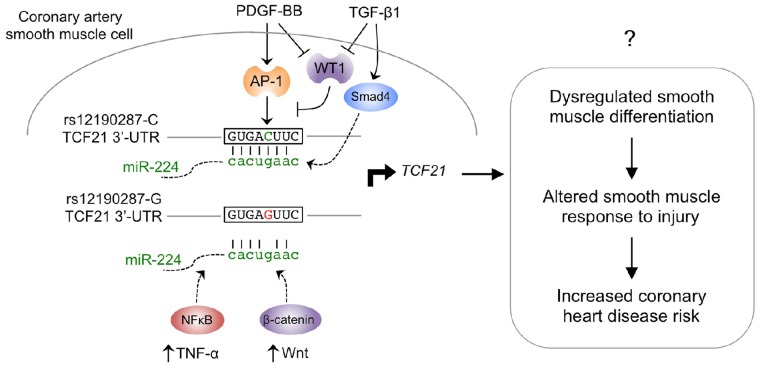
Genome-wide association studies (GWAS) have identified chromosomal loci that affect risk of coronary heart disease (CHD) independent of classical risk factors. One such association signal has been identified at 6q23.2 in both Caucasians and East Asians. The lead CHD-associated polymorphism in this region, rs12190287, resides in the 3' untranslated region (3'-UTR) of TCF21, a basic-helix-loop-helix transcription factor, and is predicted to alter the seed binding sequence for miR-224. Allelic imbalance studies in circulating leukocytes and human coronary artery smooth muscle cells (HCASMC) showed significant imbalance of the TCF21 transcript that correlated with genotype at rs12190287, consistent with this variant contributing to allele-specific expression differences. 3' UTR reporter gene transfection studies in HCASMC showed that the disease-associated C allele has reduced expression compared to the protective G allele. Kinetic analyses in vitro revealed faster RNA-RNA complex formation and greater binding of miR-224 with the TCF21 C allelic transcript. In addition, in vitro probing with Pb2+ and RNase T1 revealed structural differences between the TCF21 variants in proximity of the rs12190287 variant, which are predicted to provide greater access to the C allele for miR-224 binding. miR-224 and TCF21 expression levels were anti-correlated in HCASMC, and miR-224 modulates the transcriptional response of TCF21 to transforming growth factor-β (TGF-β) and platelet derived growth factor (PDGF) signaling in an allele-specific manner. Lastly, miR-224 and TCF21 were localized in human coronary artery lesions and anti-correlated during atherosclerosis. Together, these data suggest that miR-224 interaction with the TCF21 transcript contributes to allelic imbalance of this gene, thus partly explaining the genetic risk for coronary heart disease associated at 6q23.2. These studies implicating rs12190287 in the miRNA-dependent regulation of TCF21, in conjunction with previous studies showing that this variant modulates transcriptional regulation through activator protein 1 (AP-1), suggests a unique bimodal level of complexity previously unreported for disease-associated variants.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




References
-
- Hidai H, Bardales R, Goodwin R, Quertermous T, Quertermous EE (1998) Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech Dev 73: 33–43. - PubMed
-
- Lu J, Richardson JA, Olson EN (1998) Capsulin: a novel bHLH transcription factor expressed in epicardial progenitors and mesenchyme of visceral organs. Mech Dev 73: 23–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
