

Comparison of somatic mutation calling methods in amplicon and whole exome sequence data
- PMID: 24678773
- PMCID: PMC3986649
- DOI: 10.1186/1471-2164-15-244
Comparison of somatic mutation calling methods in amplicon and whole exome sequence data
Abstract
Background: High-throughput sequencing is rapidly becoming common practice in clinical diagnosis and cancer research. Many algorithms have been developed for somatic single nucleotide variant (SNV) detection in matched tumor-normal DNA sequencing. Although numerous studies have compared the performance of various algorithms on exome data, there has not yet been a systematic evaluation using PCR-enriched amplicon data with a range of variant allele fractions. The recently developed gold standard variant set for the reference individual NA12878 by the NIST-led "Genome in a Bottle" Consortium (NIST-GIAB) provides a good resource to evaluate admixtures with various SNV fractions.
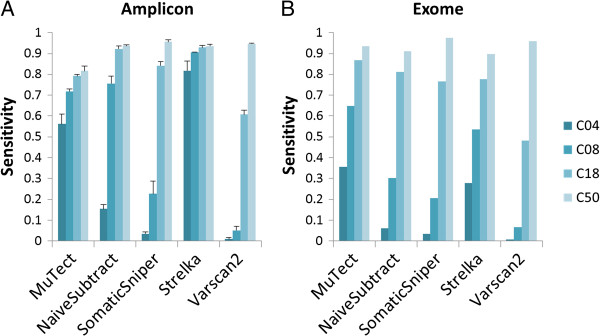
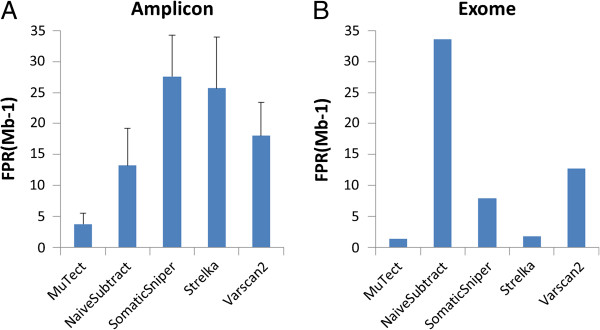
Results: Using the NIST-GIAB gold standard, we compared the performance of five popular somatic SNV calling algorithms (GATK UnifiedGenotyper followed by simple subtraction, MuTect, Strelka, SomaticSniper and VarScan2) for matched tumor-normal amplicon and exome sequencing data.
Conclusions: We demonstrated that the five commonly used somatic SNV calling methods are applicable to both targeted amplicon and exome sequencing data. However, the sensitivities of these methods vary based on the allelic fraction of the mutation in the tumor sample. Our analysis can assist researchers in choosing a somatic SNV calling method suitable for their specific needs.
Figures


References
-
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, Zhang Q, Lin L, O'Laughlin MD, McMichael JF, Delehaunty KD, Fulton LA, Magrini VJ, McGrath SD, Demeter RT, Vickery TL. et al.The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;15:264–278. doi: 10.1016/j.cell.2012.06.023. - DOI - PMC - PubMed
-
- Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, Kela I, Hopmans ES, Myklebust JH, Ji H, Plevritis SK, Levy R, Alizadeh AA. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;15:1604–1611. doi: 10.1182/blood-2012-09-457283. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
