

The immunocytokine NHS-IL12 as a potential cancer therapeutic
- PMID: 24681847
- PMCID: PMC4039112
- DOI: 10.18632/oncotarget.1853
The immunocytokine NHS-IL12 as a potential cancer therapeutic
Abstract
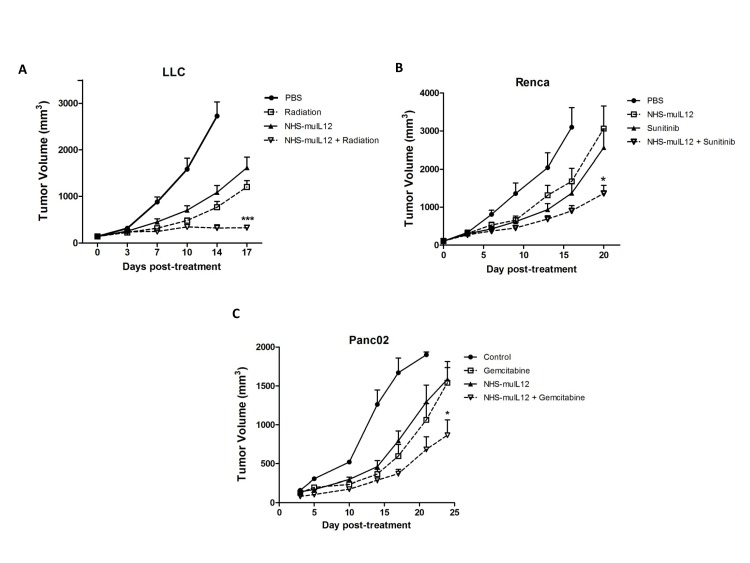
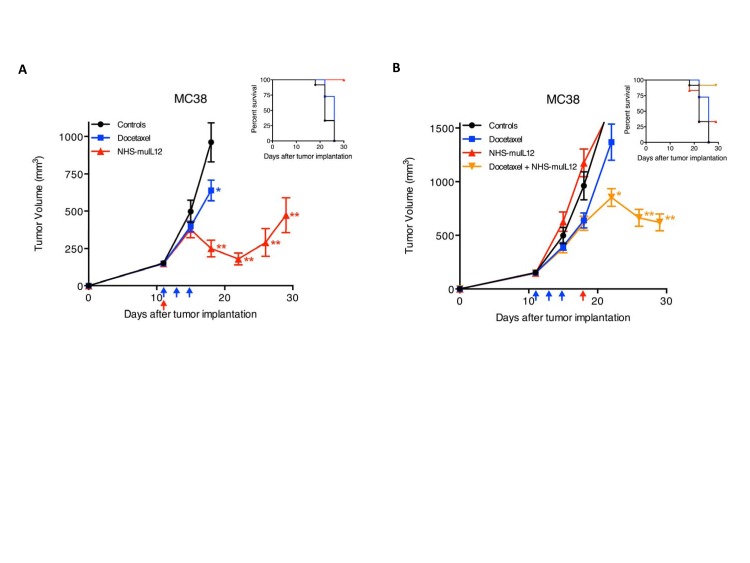
Targeted delivery of IL-12 might turn this cytokine into a safer, more effective cancer therapeutic. Here we describe a novel immunocytokine, NHS-IL12, consisting of two molecules of IL-12 fused to a tumor necrosis-targeting human IgG1 (NHS76). The addition of the human IgG1 moiety resulted in a longer plasma half-life of NHS-IL12 than recombinant IL-12, and a selective targeting to murine tumors in vivo. Data from both in vitro assays using human PBMCs and in vivo primate studies showed that IFN-gamma production by immune cells is attenuated following treatment with the immunocytokine, suggesting an improved toxicity profile than seen with recombinant IL-12 alone. NHS-IL12 was superior to recombinant IL-12 when evaluated as an anti-tumor agent in three murine tumor models. Mechanistic studies utilizing immune cell subset-depleting antibodies, flow cytometric methods, and in vitro cytotoxicity and ELISA assays all indicated that the anti-tumor effects of NHS-IL12 were primarily CD8+ T cell-dependent and likely IL-12-mediated. Combining NHS-IL12 treatment with a cancer vaccine, radiation, or chemotherapy resulted in greater anti-tumor effects than each individual therapy alone. These preclinical findings provide a rationale for the clinical testing of this immunocytokine, both as a single agent and in combination with vaccines, radiation and chemotherapy.
Conflict of interest statement
RT, GK, WG, AB, BN, YL, and HS are employees of EMD Serono. The other authors do not have any potential conflicts of interest to disclose.
Figures

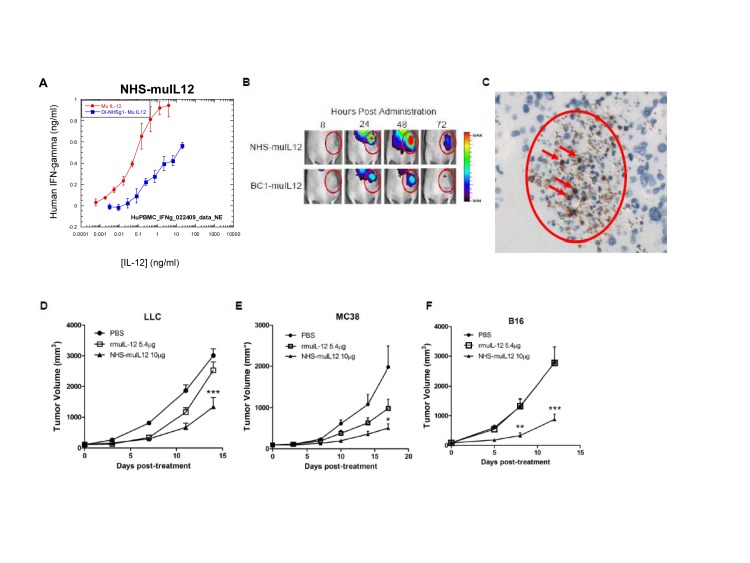
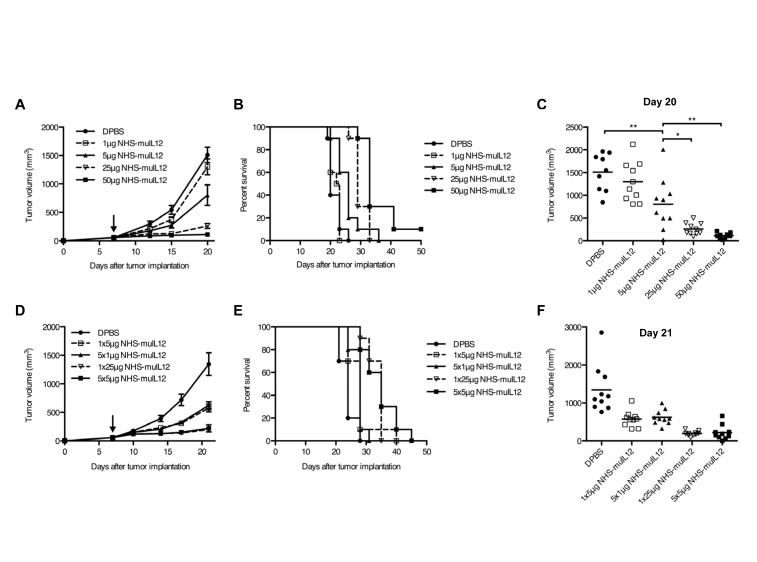
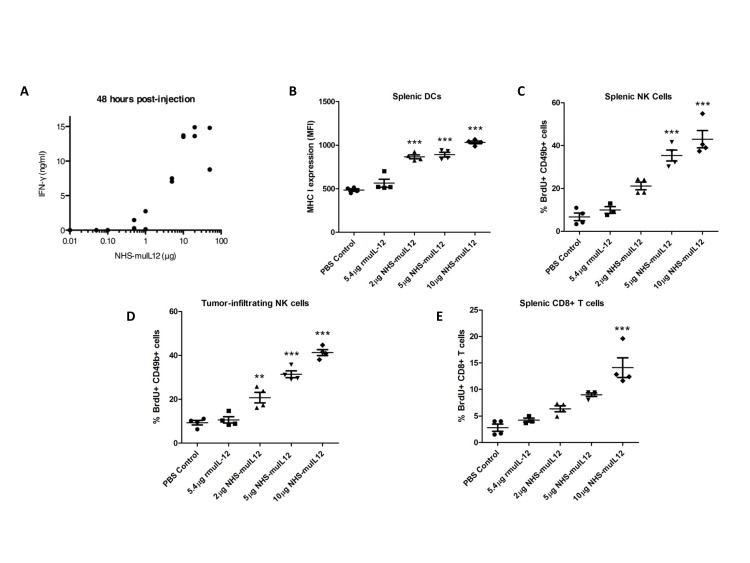
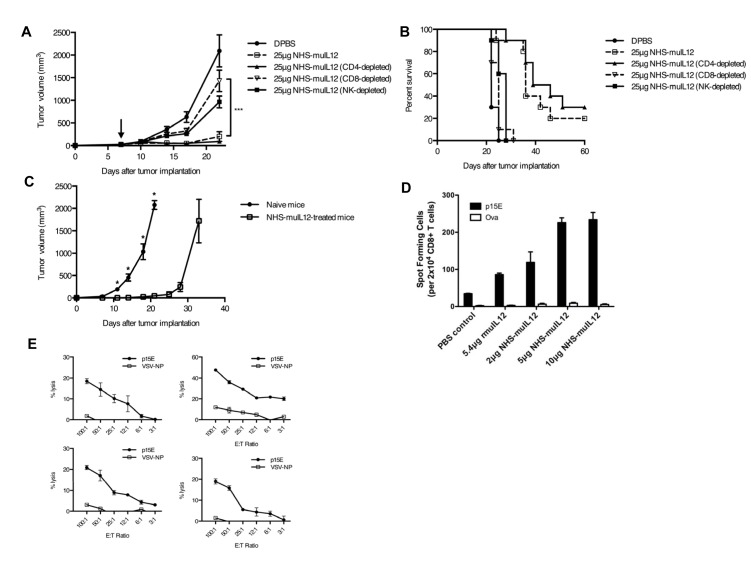

References
-
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. - PubMed
-
- Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, Romagnani S. Natural killer cell stimulatory factor (Interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med. 1993;177:1199–204. - PMC - PubMed
-
- Gately MK, Desai BB, Wolitzky AG, Quinn PM, Dwyer CM, Podlaski FJ, Familletti PC, Sinigaglia F, Chizonnite R, Gubler U, Stern AS. Regulation of human lymphocyte proliferation by a heterodimeric cytokine, IL-12 (cytotoxic lymphocyte maturation factor) J Immunol. 1991;147:874–82. - PubMed
-
- Perussia B, Chan SH, D'Andrea A, Tsuji K, Santoli D, Pospisil M, Young D, Wolf SF, Trinchieri G. Natural killer (NK) cell stimulatory factor or IL-12 has differential effects on the proliferation of TCR-αβ+, TCR-γδ+ T lymphocytes, and NK cells. J Immunol. 1992;149:3495–502. - PubMed
-
- Wong HL, Wilson DE, Jenson JC, Familletti PC, Stremlo DL, Gately MK. Characterization of a factor(s) which synergizes with recombinant interleukin 2 in promoting allogeneic human cytolytic T-lymphocyte responses in vitro. Cell Immunol. 1988;111:39–54. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
