

Genome-wide analysis of wild-type Epstein-Barr virus genomes derived from healthy individuals of the 1,000 Genomes Project
- PMID: 24682154
- PMCID: PMC4104767
- DOI: 10.1093/gbe/evu054
Genome-wide analysis of wild-type Epstein-Barr virus genomes derived from healthy individuals of the 1,000 Genomes Project
Abstract
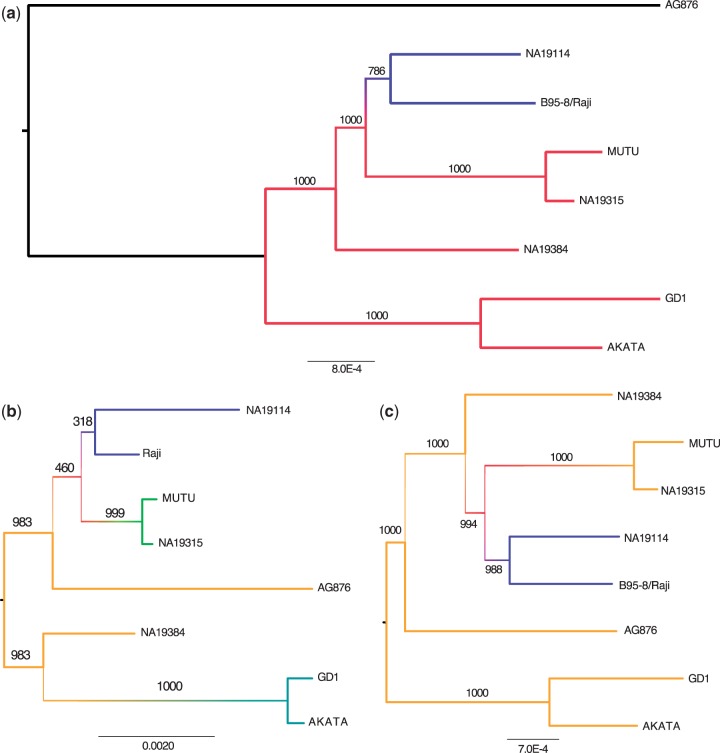
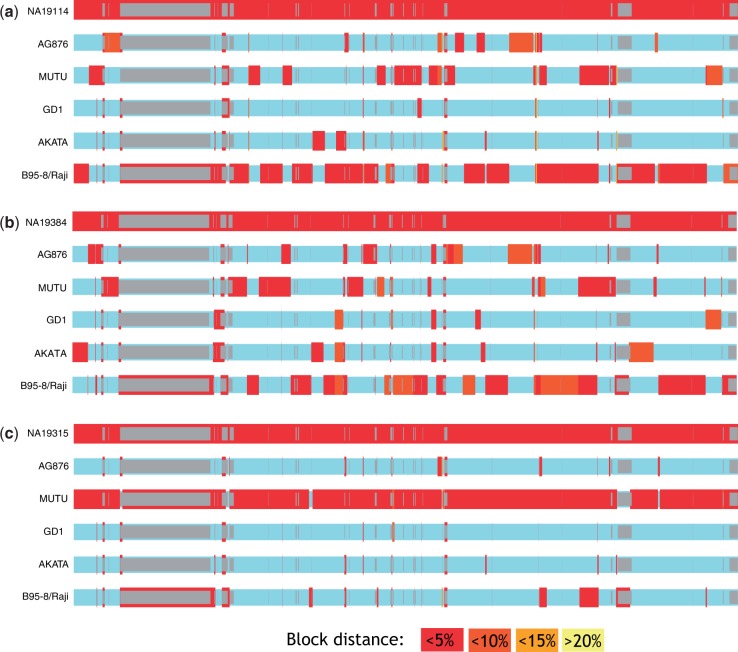
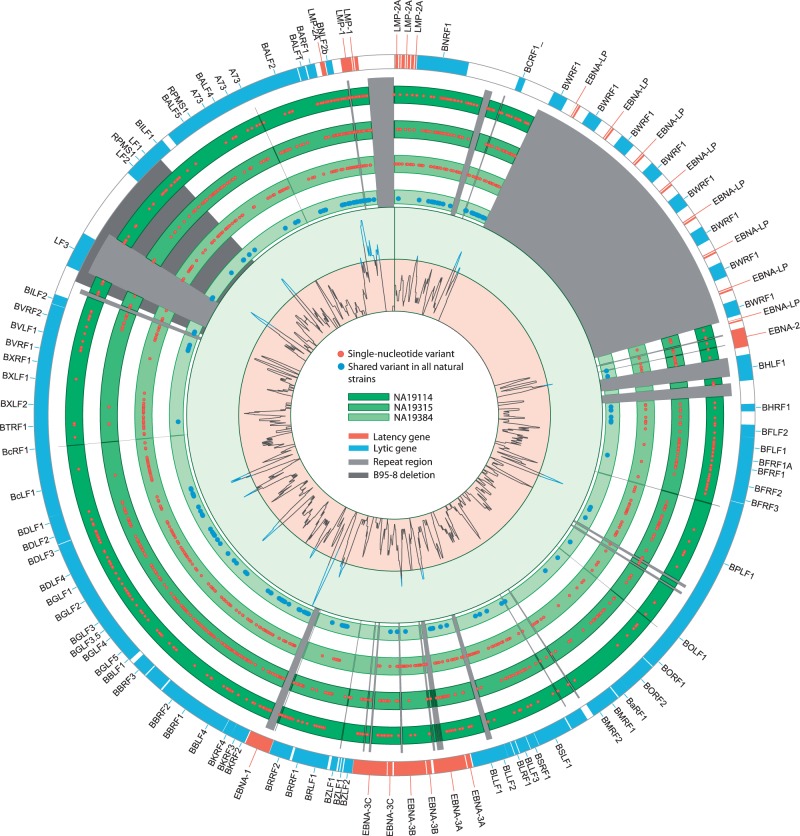
Most people in the world (∼90%) are infected by the Epstein-Barr virus (EBV), which establishes itself permanently in B cells. Infection by EBV is related to a number of diseases including infectious mononucleosis, multiple sclerosis, and different types of cancer. So far, only seven complete EBV strains have been described, all of them coming from donors presenting EBV-related diseases. To perform a detailed comparative genomic analysis of EBV including, for the first time, EBV strains derived from healthy individuals, we reconstructed EBV sequences infecting lymphoblastoid cell lines (LCLs) from the 1000 Genomes Project. As strain B95-8 was used to transform B cells to obtain LCLs, it is always present, but a specific deletion in its genome sets it apart from natural EBV strains. After studying hundreds of individuals, we determined the presence of natural EBV in at least 10 of them and obtained a set of variants specific to wild-type EBV. By mapping the natural EBV reads into the EBV reference genome (NC007605), we constructed nearly complete wild-type viral genomes from three individuals. Adding them to the five disease-derived EBV genomic sequences available in the literature, we performed an in-depth comparative genomic analysis. We found that latency genes harbor more nucleotide diversity than lytic genes and that six out of nine latency-related genes, as well as other genes involved in viral attachment and entry into host cells, packaging, and the capsid, present the molecular signature of accelerated protein evolution rates, suggesting rapid host-parasite coevolution.
Keywords: EBV; Illumina reads; human herpesvirus 4; recombination; selection; whole-genome analysis.
Figures
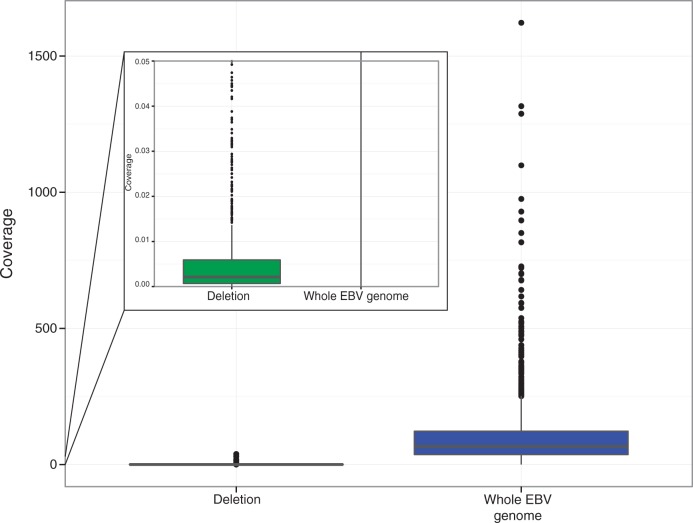
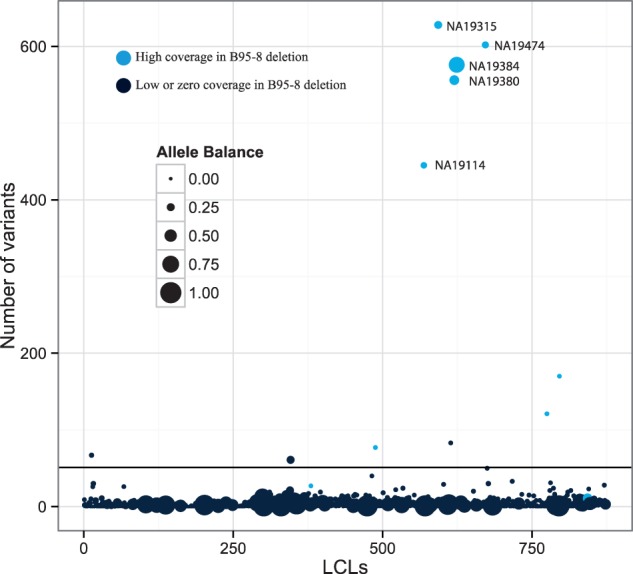
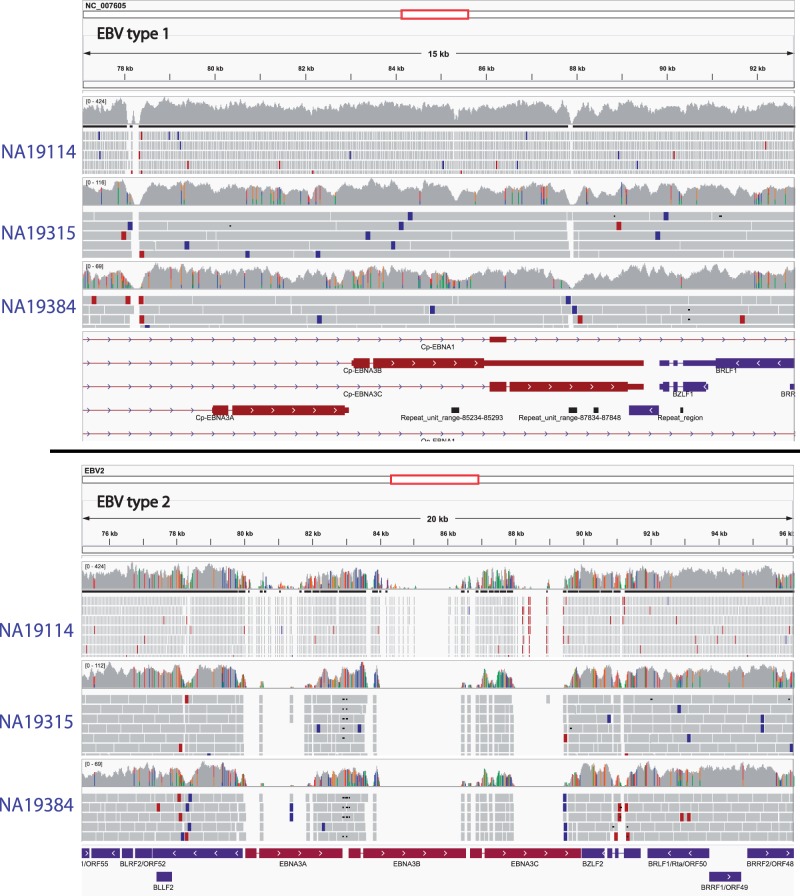
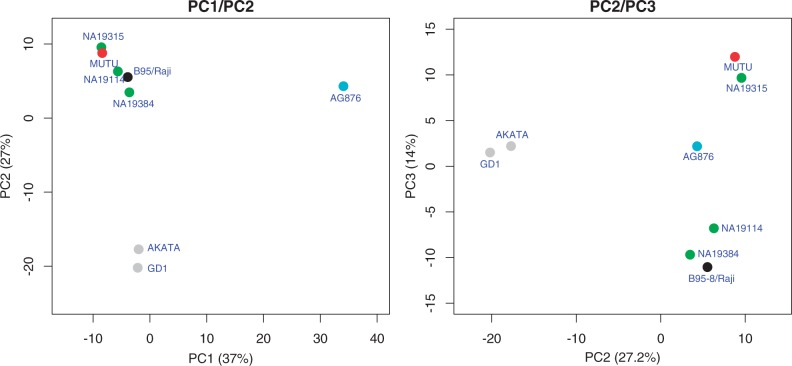
References
-
- Aitken C, Sengupta SK, Aedes C, Moss DJ, Sculley TB. Heterogeneity within the Epstein-Barr virus nuclear antigen 2 gene in different strains of Epstein-Barr virus. J Gen Virol. 1994;75(Pt 1):95–100. - PubMed
-
- Al-Mozaini M, et al. Epstein-Barr virus BART gene expression. J Gen Virol. 2009;90:307–316. - PubMed
-
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001;18:1585–15892. - PubMed
-
- Ascherio A, Munger KL. Epstein-barr virus infection and multiple sclerosis: a review. J Neuroimmune Pharmacol. 2010;5:271–277. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
