

Proteomic identification of monoclonal antibodies from serum
- PMID: 24684310
- PMCID: PMC4033631
- DOI: 10.1021/ac4037679
Proteomic identification of monoclonal antibodies from serum
Abstract
Characterizing the in vivo dynamics of the polyclonal antibody repertoire in serum, such as that which might arise in response to stimulation with an antigen, is difficult due to the presence of many highly similar immunoglobulin proteins, each specified by distinct B lymphocytes. These challenges have precluded the use of conventional mass spectrometry for antibody identification based on peptide mass spectral matches to a genomic reference database. Recently, progress has been made using bottom-up analysis of serum antibodies by nanoflow liquid chromatography/high-resolution tandem mass spectrometry combined with a sample-specific antibody sequence database generated by high-throughput sequencing of individual B cell immunoglobulin variable domains (V genes). Here, we describe how intrinsic features of antibody primary structure, most notably the interspersed segments of variable and conserved amino acid sequences, generate recurring patterns in the corresponding peptide mass spectra of V gene peptides, greatly complicating the assignment of correct sequences to mass spectral data. We show that the standard method of decoy-based error modeling fails to account for the error introduced by these highly similar sequences, leading to a significant underestimation of the false discovery rate. Because of these effects, antibody-derived peptide mass spectra require increased stringency in their interpretation. The use of filters based on the mean precursor ion mass accuracy of peptide-spectrum matches is shown to be particularly effective in distinguishing between "true" and "false" identifications. These findings highlight important caveats associated with the use of standard database search and error-modeling methods with nonstandard data sets and custom sequence databases.
Figures
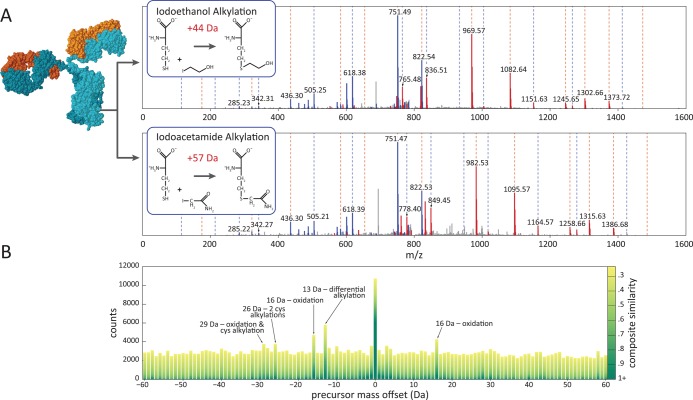
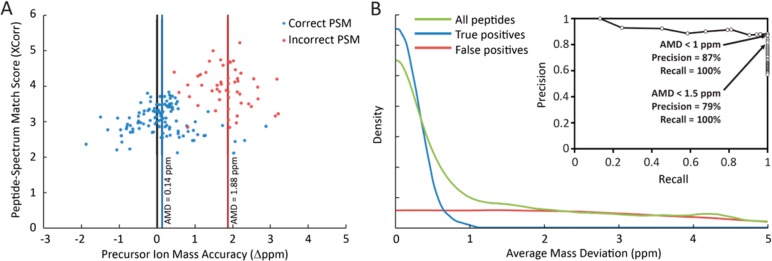
References
-
- Poulsen T. R.; Meijer P. J.; Jensen A.; Nielsen L. S.; Andersen P. S. J. Immunol 2007, 179, 3841–3850. - PubMed
-
- Murphy K.; Travers P.; Walport M.; Janeway C.. Janeway′s immunobiology, 8th ed.; Garland Science: New York, 2012; p xix, 868 p.
-
- Tarlinton D.; Good-Jacobson K. Science 2013, 341, 1205–1211. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
