

Cystic fibrosis: an inherited disease affecting mucin-producing organs
- PMID: 24685676
- PMCID: PMC4449140
- DOI: 10.1016/j.biocel.2014.03.011
Cystic fibrosis: an inherited disease affecting mucin-producing organs
Abstract
Our current understanding of cystic fibrosis (CF) has revealed that the biophysical properties of mucus play a considerable role in the pathogenesis of the disease in view of the fact that most mucus-producing organs are affected in CF patients. In this review, we discuss the potential causal relationship between altered cystic fibrosis transmembrane conductance regulator (CFTR) function and the production of mucus with abnormal biophysical properties in the intestine and lungs, highlighting what has been learned from cell cultures and animal models that mimic CF pathogenesis. A similar cascade of events, including mucus obstruction, infection and inflammation, is common to all epithelia affected by impaired surface hydration. Hence, the main structural components of mucus, namely the polymeric, gel-forming mucins, are critical to the onset of the disease. Defective CFTR leads to epithelial surface dehydration, altered pH/electrolyte composition and mucin concentration. Further, it can influence mucin transition from the intracellular to extracellular environment, potentially resulting in aberrant mucus gel formation. While defective HCO3(-) production has long been identified as a feature of CF, it has only recently been considered as a key player in the transition phase of mucins. We conclude by examining the influence of mucins on the biophysical properties of CF sputum and discuss existing and novel therapies aimed at removing mucus from the lungs. This article is part of a Directed Issue entitled: Cystic Fibrosis: From o-mics to cell biology, physiology, and therapeutic advances.
Keywords: CF; CFTR; Mucin; Mucus; Pathogenesis.
Copyright © 2014 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
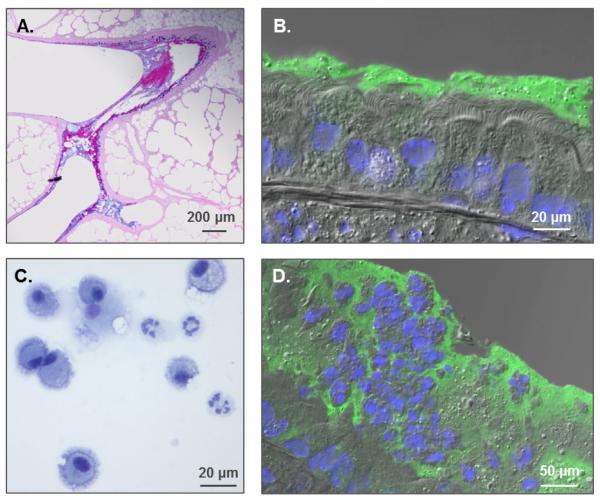
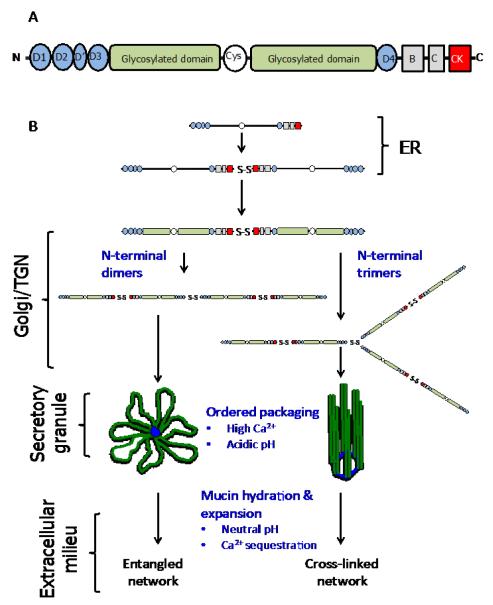
References
-
- Altes T, Johnson MA, Miller GW, Mugler JP, Flors L, Mata J, et al. Hyperpolarized gas MRI of Ivacaftor therapy in persons with cystic fibrosis and the G551D-CFTR mutation. Pediatric Pulmonology. 2012;(Supplement 35):291.
-
- Asker N, Axelsson MA, Olofsson SO, Hansson GC. Dimerization of the human MUC2 mucin in the endoplasmic reticulum is followed by a N-glycosylation-dependent transfer of the mono- and dimers to the Golgi apparatus. J Biol Chem. 1998a;273:18857–63. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
