

Pre-exposure to adenosine, acting via A(2A) receptors on endothelial cells, alters the protein kinase A dependence of adenosine-induced dilation in skeletal muscle resistance arterioles
- PMID: 24687580
- PMCID: PMC4080939
- DOI: 10.1113/jphysiol.2013.265835
Pre-exposure to adenosine, acting via A(2A) receptors on endothelial cells, alters the protein kinase A dependence of adenosine-induced dilation in skeletal muscle resistance arterioles
Abstract
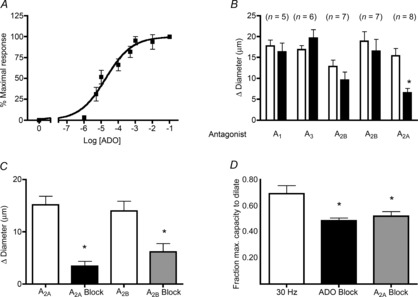
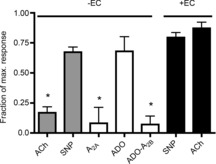
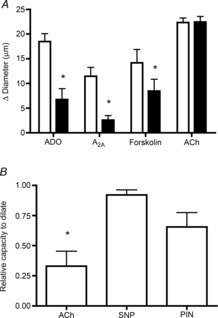
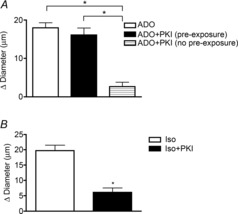
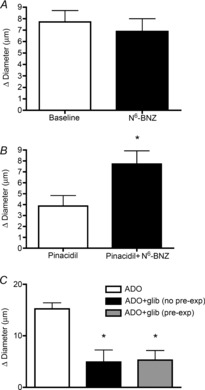
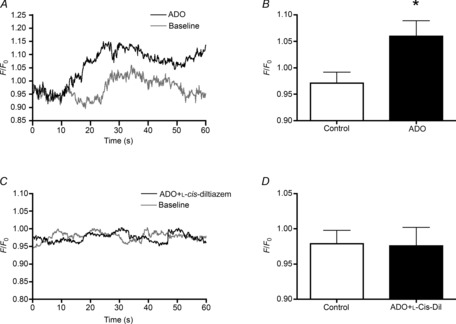
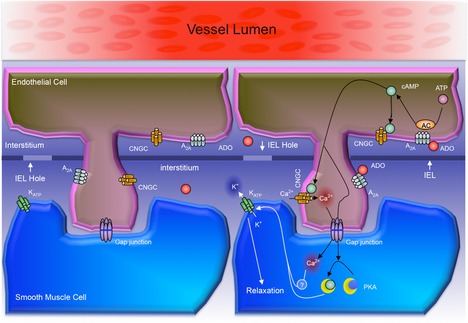
Adenosine (ADO) is an endogenous vasodilatory purine widely recognized to be a significant contributor to functional hyperaemia. Despite this, many aspects of the mechanisms by which ADO induces dilation in small resistance arterioles are not established, or appear contradictory. These include: identification of the primary receptor subtype; its location on endothelial (EC) or vascular smooth muscle cells; whether ADO acts on KATP channels in these resistance vessels; and the contribution of cAMP/protein kinase A (PKA) signalling to the response. In intravital microscopy studies of intact or EC-denuded skeletal muscle arterioles, we show that ADO acts via A2A receptors located on ECs to produce vasodilation via activation of KATP channels located on vascular smooth muscle cells. Importantly, we found that the signalling pathway involves cAMP as expected, but that a requirement for PKA activation is demonstrable only if the vessel is not pre-exposed to ADO. That is, PKA-dependent signalling varies with pre-exposure to ADO. Further, we show that PKA activation alone is not sufficient to dilate these arterioles; an additional EC calcium-dependent signalling mechanism is required for vasodilation to ADO. The ability of arterioles in situ to respond to occupancy of a specific receptor by utilizing different cell signalling pathways under different conditions to produce the same response allows the arteriole to respond to key homeostatic requirements using more than a single signalling mechanism. Clearly, this is likely to be physiologically advantageous, but the role for this signalling flexibility in the integrated arteriolar response that underlies functional hyperaemia will require further exploration.
© 2014 The Authors. The Journal of Physiology © 2014 The Physiological Society.
Figures


References
-
- Banitt PF, Smits P, Williams SB, Ganz P, Creager MA. Activation of ATP-sensitive potassium channels contributes to reactive hyperemia in humans. Am J Physiol Heart Circ Physiol. 1996;271:H1594–H1598. - PubMed
-
- Bockman EL, Berne RM, Rubio R. Adenosine and active hyperemia in dog skeletal muscle. Am J Physiol. 1976;230:1531–1537. - PubMed
-
- Carroll MA, Doumad AB, Li J, Cheng MK, Falck JR, McGiff JC. Adenosine2A receptor vasodilation of rat preglomerular microvessels is mediated by EETs that activate the cAMP/PKA pathway. Am J Physiol Renal Physiol. 2006;291:F155–F161. - PubMed
-
- Cheng KT, Leung YK, Shen B, Kwok YC, Wong CO, Kwan HY, Man YB, Ma X, Huang Y, Yao X. CNGA2 channels mediate adenosine-induced Ca2+ influx in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:913–918. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources