

Biochemical and biophysical characterization of recombinant yeast proteasome maturation factor ump1
- PMID: 24688736
- PMCID: PMC3962104
- DOI: 10.5936/csbj.201304006
Biochemical and biophysical characterization of recombinant yeast proteasome maturation factor ump1
Abstract
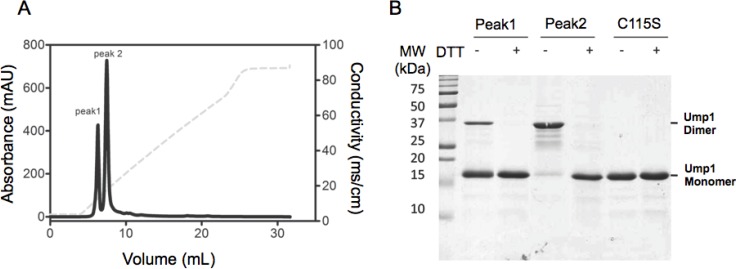
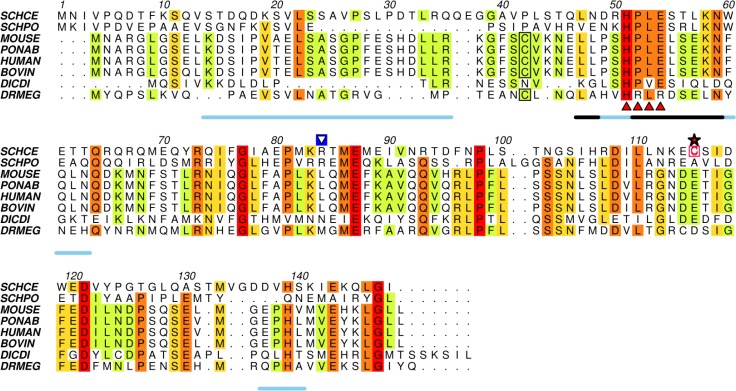
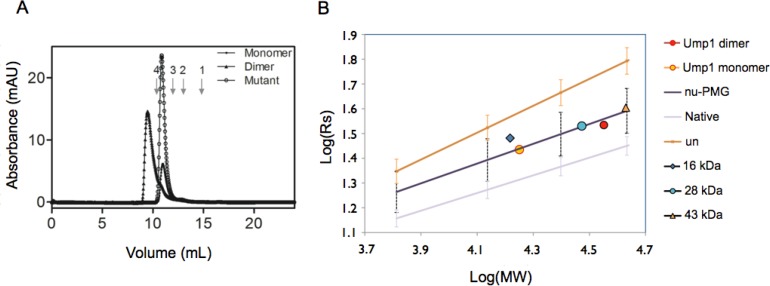
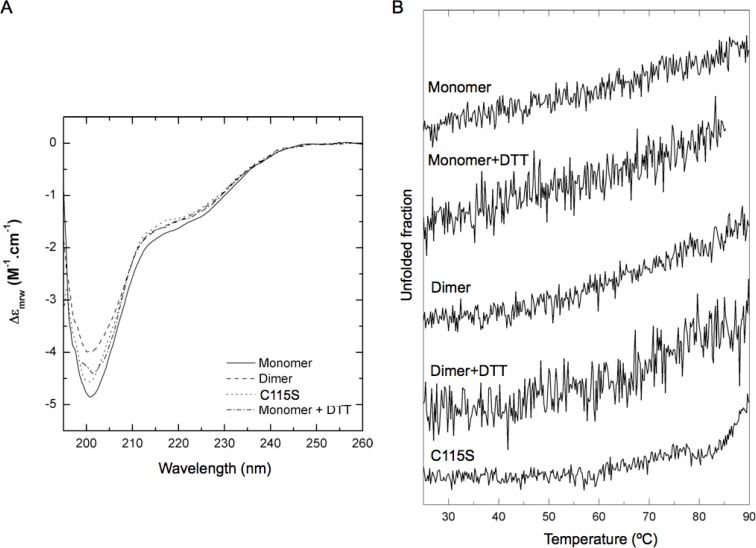
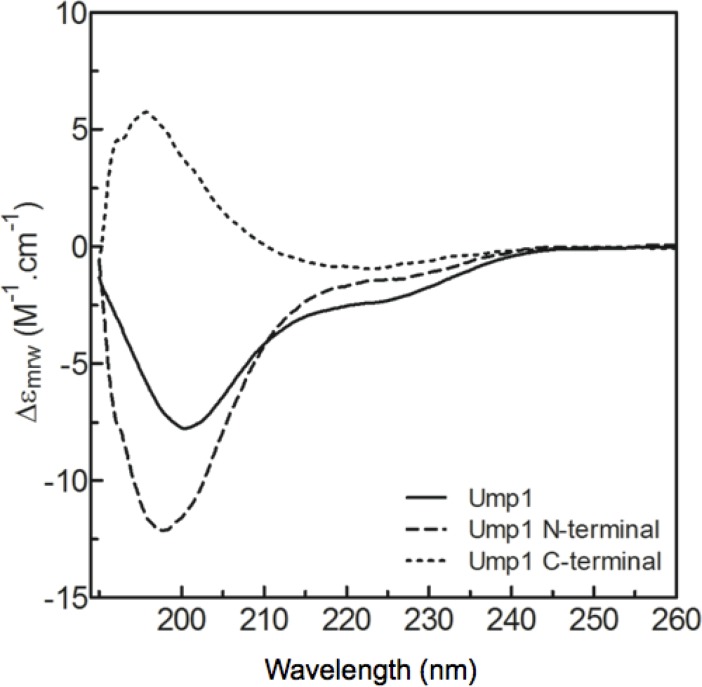
Protein degradation is essential for maintaining cellular homeostasis. The proteasome is the central enzyme responsible for non-lysosomal protein degradation in eukaryotic cells. Although proteasome assembly is not yet completely understood, a number of cofactors required for proper assembly and maturation have been identified. Ump is a short-lived maturation factor required for the efficient biogenesis of the 20S proteasome. Upon the association of the two precursor complexes, Ump is encased and is rapidly degraded after the proteolytic sites in the interior of the nascent proteasome are activated. In order to further understand the mechanisms behind proteasomal maturation, we expressed and purified yeast Ump in E. coli for biophysical and structural analysis. We show that recombinant Ump is purified as a mixture of different oligomeric species and that oligomerization is mediated by intermolecular disulfide bond formation involving the only cysteine residue present in the protein. Furthermore, a combination of bioinformatic, biochemical and structural analysis revealed that Ump shows characteristics of an intrinsically disordered protein, which might become structured only upon interaction with the proteasome subunits.
Keywords: Circular dichroism; dynamic light scattering; intrinsically disordered; protein structure.
Figures
References
-
- Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ (2009) Catalytic mechanism and assembly of the proteasome. Chem Rev 109: 1509–1536 - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases