

Deciphering intrafamilial phenotypic variability by exome sequencing in a Bardet-Biedl family
- PMID: 24689075
- PMCID: PMC3960054
- DOI: 10.1002/mgg3.50
Deciphering intrafamilial phenotypic variability by exome sequencing in a Bardet-Biedl family
Abstract
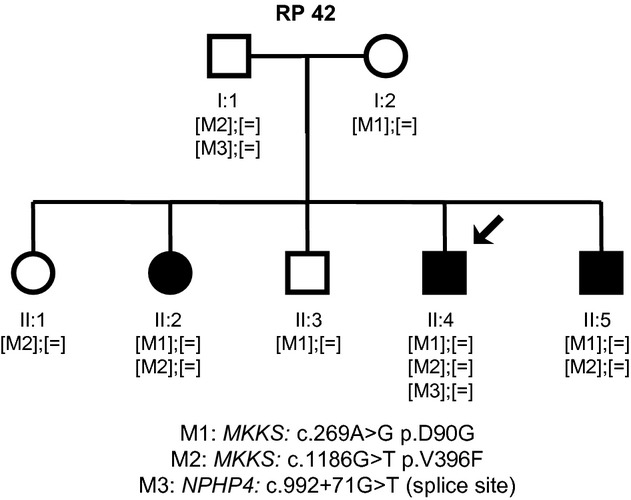
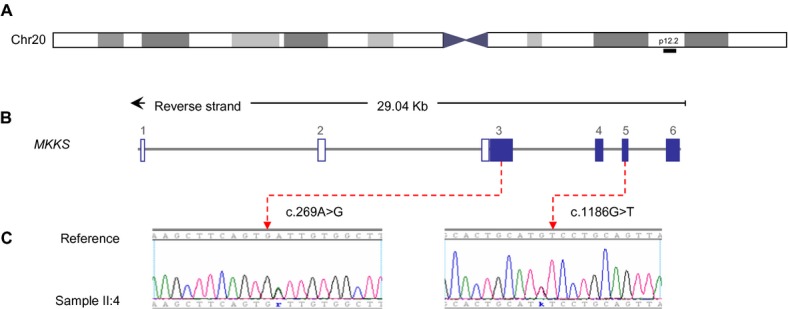
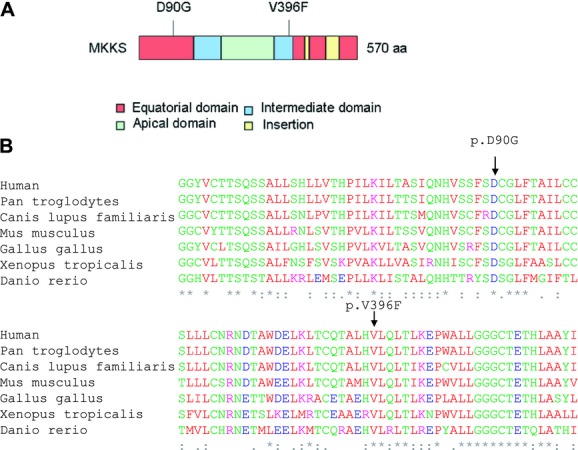
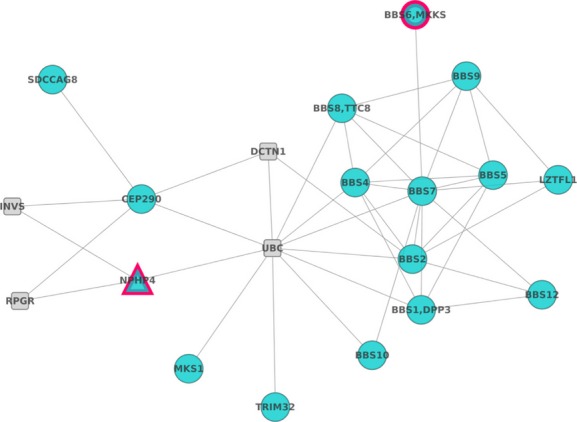
Bardet-Biedl syndrome (BBS) is a model ciliopathy characterized by a wide range of clinical variability. The heterogeneity of this condition is reflected in the number of underlying gene defects and the epistatic interactions between the proteins encoded. BBS is generally inherited in an autosomal recessive trait. However, in some families, mutations across different loci interact to modulate the expressivity of the phenotype. In order to investigate the magnitude of epistasis in one BBS family with remarkable intrafamilial phenotypic variability, we designed an exome sequencing-based approach using SOLID 5500xl platform. This strategy allowed the reliable detection of the primary causal mutations in our family consisting of two novel compound heterozygous mutations in McKusick-Kaufman syndrome (MKKS) gene (p.D90G and p.V396F). Additionally, exome sequencing enabled the detection of one novel heterozygous NPHP4 variant which is predicted to activate a cryptic acceptor splice site and is only present in the most severely affected patient. Here, we provide an exome sequencing analysis of a BBS family and show the potential utility of this tool, in combination with network analysis, to detect disease-causing mutations and second-site modifiers. Our data demonstrate how next-generation sequencing (NGS) can facilitate the dissection of epistatic phenomena, and shed light on the genetic basis of phenotypic variability.
Keywords: Bardet–Biedl Syndrome; MKKS; NGS; NPHP4; intrafamilial variability.
Figures
Similar articles
-
Mutation analysis of the MKKS gene in McKusick-Kaufman syndrome and selected Bardet-Biedl syndrome patients.Hum Genet. 2002 Jun;110(6):561-7. doi: 10.1007/s00439-002-0733-3. Epub 2002 May 9. Hum Genet. 2002. PMID: 12107442
-
McKusick-Kaufman or Bardet-Biedl syndrome? A new borderline case in an Italian nonconsanguineous healthy family.Indian J Hum Genet. 2011 May;17(2):94-6. doi: 10.4103/0971-6866.86194. Indian J Hum Genet. 2011. PMID: 22090721 Free PMC article.
-
Exome sequencing identifies a novel and a recurrent BBS1 mutation in Pakistani families with Bardet-Biedl syndrome.Mol Vis. 2013;19:644-53. Epub 2013 Mar 21. Mol Vis. 2013. PMID: 23559858 Free PMC article.
-
Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney.Pediatr Nephrol. 2012 Jan;27(1):7-15. doi: 10.1007/s00467-010-1751-3. Epub 2011 Jan 19. Pediatr Nephrol. 2012. PMID: 21246219 Review.
-
The oligogenic properties of Bardet-Biedl syndrome.Hum Mol Genet. 2004 Apr 1;13 Spec No 1:R65-71. doi: 10.1093/hmg/ddh092. Epub 2004 Feb 19. Hum Mol Genet. 2004. PMID: 14976158 Review.
Cited by
-
Identification of a Novel Homozygous Missense (c.443A>T:p.N148I) Mutation in BBS2 in a Kashmiri Family with Bardet-Biedl Syndrome.Biomed Res Int. 2021 Feb 23;2021:6626015. doi: 10.1155/2021/6626015. eCollection 2021. Biomed Res Int. 2021. PMID: 33688495 Free PMC article.
-
Exome sequencing reveals novel and recurrent mutations with clinical significance in inherited retinal dystrophies.PLoS One. 2014 Dec 29;9(12):e116176. doi: 10.1371/journal.pone.0116176. eCollection 2014. PLoS One. 2014. PMID: 25544989 Free PMC article.
-
Bardet-Biedl syndrome caused by compound heterozygosity in BBS12 gene: a case report of one family with three affected members.Front Pediatr. 2023 Jul 4;11:1226595. doi: 10.3389/fped.2023.1226595. eCollection 2023. Front Pediatr. 2023. PMID: 37469681 Free PMC article.
-
Novel RP1 mutations and a recurrent BBS1 variant explain the co-existence of two distinct retinal phenotypes in the same pedigree.BMC Genet. 2014 Dec 14;15:143. doi: 10.1186/s12863-014-0143-2. BMC Genet. 2014. PMID: 25494902 Free PMC article.
-
Bardet-Biedl syndrome: Genetics, molecular pathophysiology, and disease management.Indian J Ophthalmol. 2016 Sep;64(9):620-627. doi: 10.4103/0301-4738.194328. Indian J Ophthalmol. 2016. PMID: 27853007 Free PMC article. Review.
References
-
- Badano JL, Kim JC, Hoskins BE, Lewis RA, Ansley SJ, Cutler DJ. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 2003;12:1651–1659. - PubMed
-
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006a;7:125–148. - PubMed
-
- Badano JL, Leitch CC, Ansley SJ, May-Simera H, Lawson S, Lewis RA. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006b;439:326–330. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
