

Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D
- PMID: 24694735
- PMCID: PMC4069603
- DOI: 10.1158/2159-8290.CD-13-0593
Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D
Abstract
Cancer-associated inflammation is a molecular key feature in pancreatic ductal adenocarcinoma. Oncogenic KRAS in conjunction with persistent inflammation is known to accelerate carcinogenesis, although the underlying mechanisms remain poorly understood. Here, we outline a novel pathway whereby the transcription factors NFATc1 and STAT3 cooperate in pancreatic epithelial cells to promote Kras(G12D)-driven carcinogenesis. NFATc1 activation is induced by inflammation and itself accelerates inflammation-induced carcinogenesis in Kras(G12D) mice, whereas genetic or pharmacologic ablation of NFATc1 attenuates this effect. Mechanistically, NFATc1 complexes with STAT3 for enhancer-promoter communications at jointly regulated genes involved in oncogenesis, for example, Cyclin, EGFR and WNT family members. The NFATc1-STAT3 cooperativity is operative in pancreatitis-mediated carcinogenesis as well as in established human pancreatic cancer. Together, these studies unravel new mechanisms of inflammatory-driven pancreatic carcinogenesis and suggest beneficial effects of chemopreventive strategies using drugs that are currently available for targeting these factors in clinical trials.
Significance: Our study points to the existence of an oncogenic NFATc1-STAT3 cooperativity that mechanistically links inflammation with pancreatic cancer initiation and progression. Because NFATc1-STAT3 nucleoprotein complexes control the expression of gene networks at the intersection of inflammation and cancer, our study has significant relevance for potentially managing pancreatic cancer and other inflammatory-driven malignancies.
©2014 American Association for Cancer Research.
Conflict of interest statement
The authors have no conflict of interest to declare.
Figures


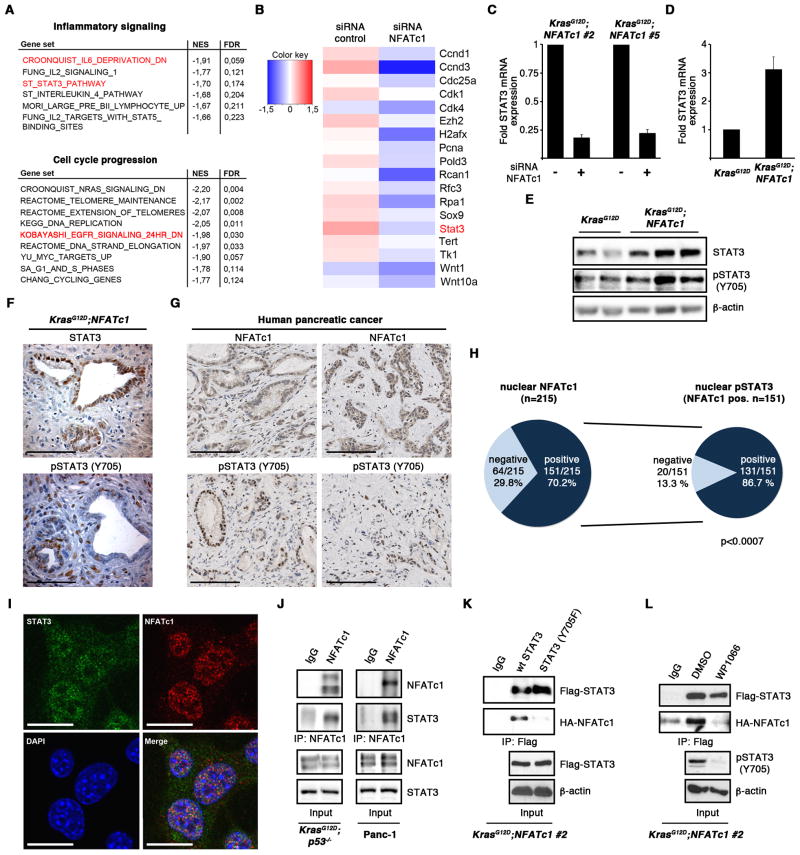
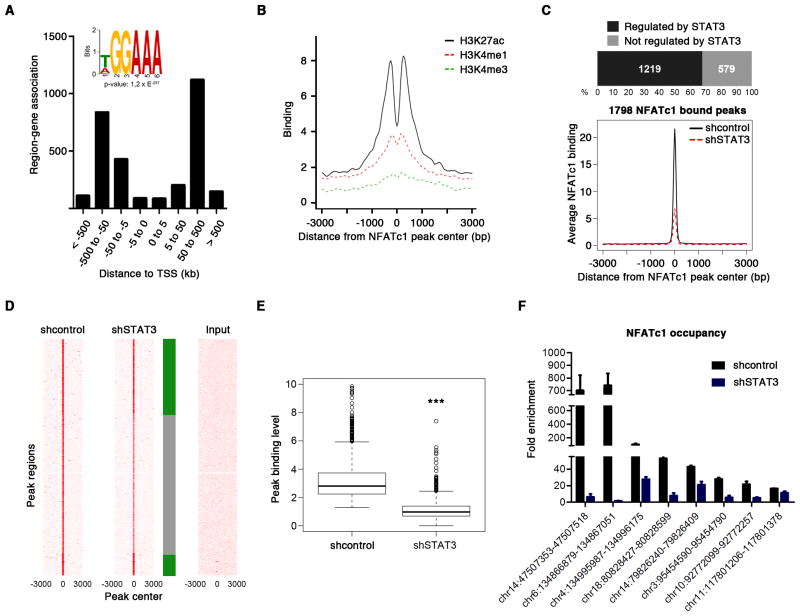

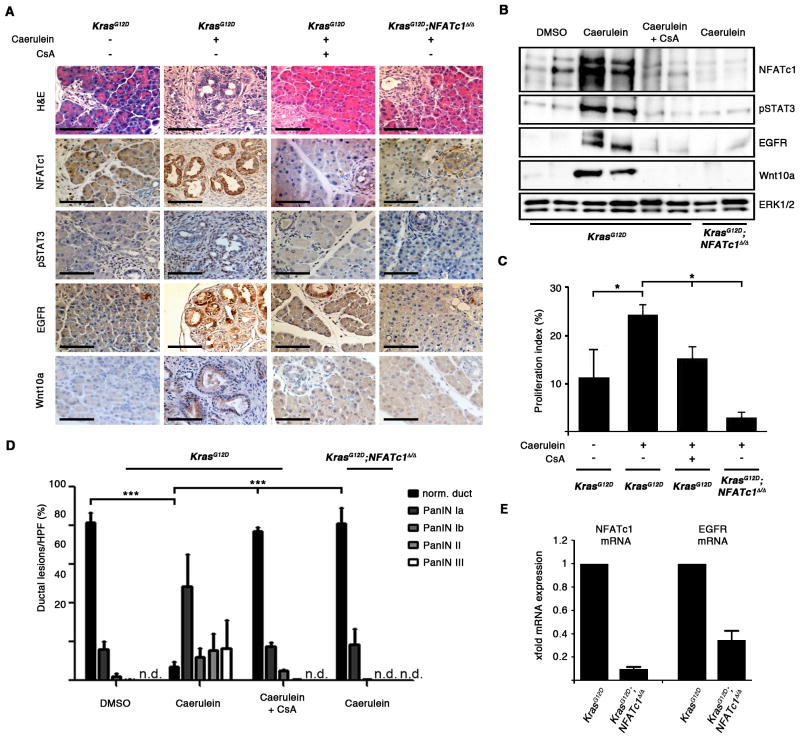
References
-
- Warshaw AL, Fernández-del Castillo C. Pancreatic carcinoma. N Engl J Med. 1992;326:455–65. - PubMed
-
- Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–17. - PubMed
-
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
