

Runx1 regulation of Pu.1 corepressor/coactivator exchange identifies specific molecular targets for leukemia differentiation therapy
- PMID: 24695740
- PMCID: PMC4031539
- DOI: 10.1074/jbc.M114.562447
Runx1 regulation of Pu.1 corepressor/coactivator exchange identifies specific molecular targets for leukemia differentiation therapy
Abstract
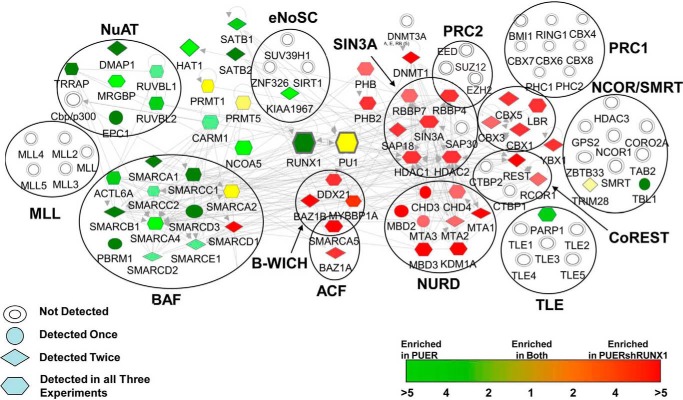
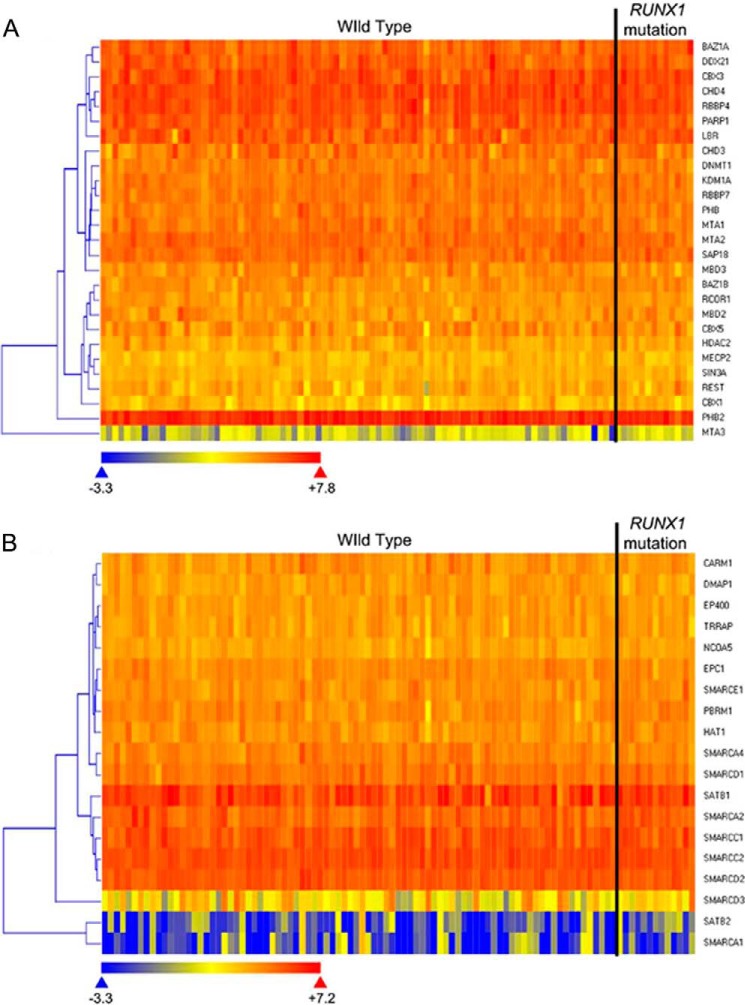
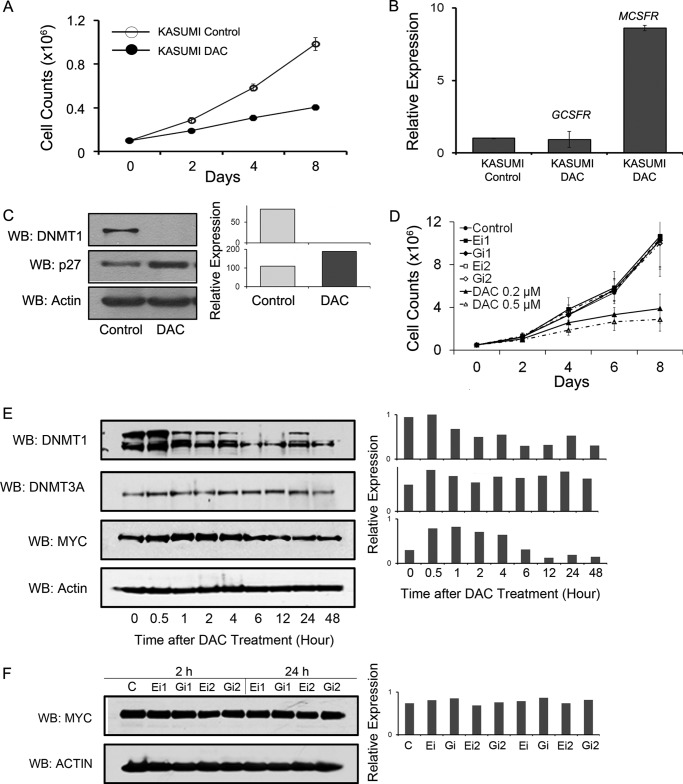
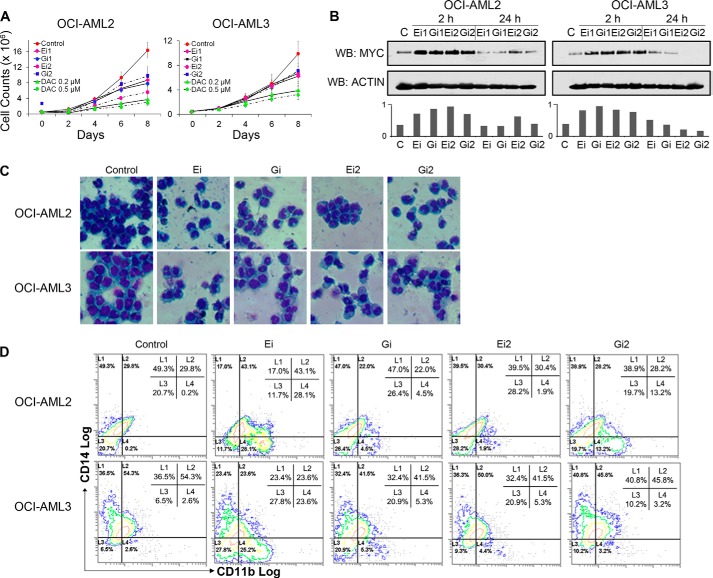
Gene activation requires cooperative assembly of multiprotein transcription factor-coregulator complexes. Disruption to cooperative assemblage could underlie repression of tumor suppressor genes in leukemia cells. Mechanisms of cooperation and its disruption were therefore examined for PU.1 and RUNX1, transcription factors that cooperate to activate hematopoietic differentiation genes. PU.1 is highly expressed in leukemia cells, whereas RUNX1 is frequently inactivated by mutation or translocation. Thus, coregulator interactions of Pu.1 were examined by immunoprecipitation coupled with tandem mass spectrometry/Western blot in wild-type and Runx1-deficient hematopoietic cells. In wild-type cells, the NuAT and Baf families of coactivators coimmunoprecipitated with Pu.1. Runx1 deficiency produced a striking switch to Pu.1 interaction with the Dnmt1, Sin3A, Nurd, CoRest, and B-Wich corepressor families. Corepressors of the Polycomb family, which are frequently inactivated by mutation or deletion in myeloid leukemia, did not interact with Pu.1. The most significant gene ontology association of Runx1-Pu.1 co-bound genes was with macrophages, therefore, functional consequences of altered corepressor/coactivator exchange were examined at Mcsfr, a key macrophage differentiation gene. In chromatin immunoprecipitation analyses, high level Pu.1 binding to the Mcsfr promoter was not decreased by Runx1 deficiency. However, the Pu.1-driven shift from histone repression to activation marks at this locus, and terminal macrophage differentiation, were substantially diminished. DNMT1 inhibition, but not Polycomb inhibition, in RUNX1-translocated leukemia cells induced terminal differentiation. Thus, RUNX1 and PU.1 cooperate to exchange corepressors for coactivators, and the specific corepressors recruited to PU.1 as a consequence of RUNX1 deficiency could be rational targets for leukemia differentiation therapy.
Keywords: Chromatin Modifying Enzymes; Enhanceosome; Enzyme Inhibitors; Epigenetics; Leukemogenesis; Protein Complexes; Protein Interactome; Transcription Coactivators; Transcription Regulation; Transcription Repressor.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures

References
-
- Rosenfeld M. G., Lunyak V. V., Glass C. K. (2006) Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 20, 1405–1428 - PubMed
-
- Perissi V., Jepsen K., Glass C. K., Rosenfeld M. G. (2010) Deconstructing repression: evolving models of co-repressor action. Nat. Rev. Genet. 11, 109–123 - PubMed
-
- Ram O., Goren A., Amit I., Shoresh N., Yosef N., Ernst J., Kellis M., Gymrek M., Issner R., Coyne M., Durham T., Zhang X., Donaghey J., Epstein C. B., Regev A., Bernstein B. E. (2011) Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147, 1628–1639 - PMC - PubMed
-
- Niimi H., Harada H., Harada Y., Ding Y., Imagawa J., Inaba T., Kyo T., Kimura A. (2006) Hyperactivation of the RAS signaling pathway in myelodysplastic syndrome with AML1/RUNX1 point mutations. Leukemia 20, 635–644 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
