

Platelets and cancer: a casual or causal relationship: revisited
- PMID: 24696047
- PMCID: PMC4186918
- DOI: 10.1007/s10555-014-9498-0
Platelets and cancer: a casual or causal relationship: revisited
Abstract
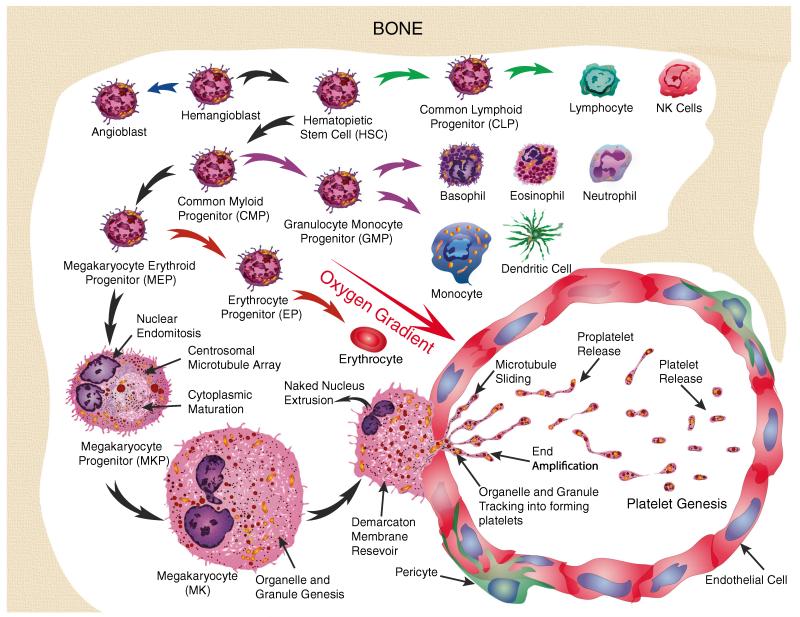
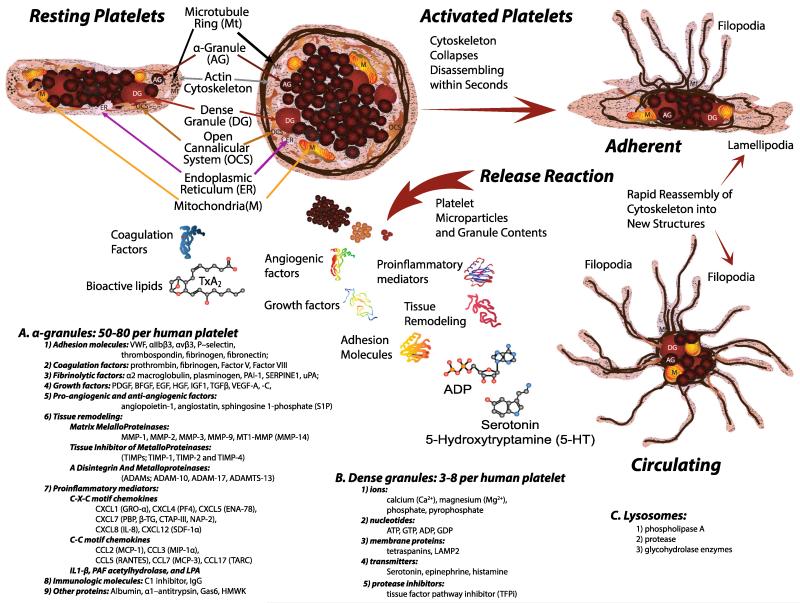
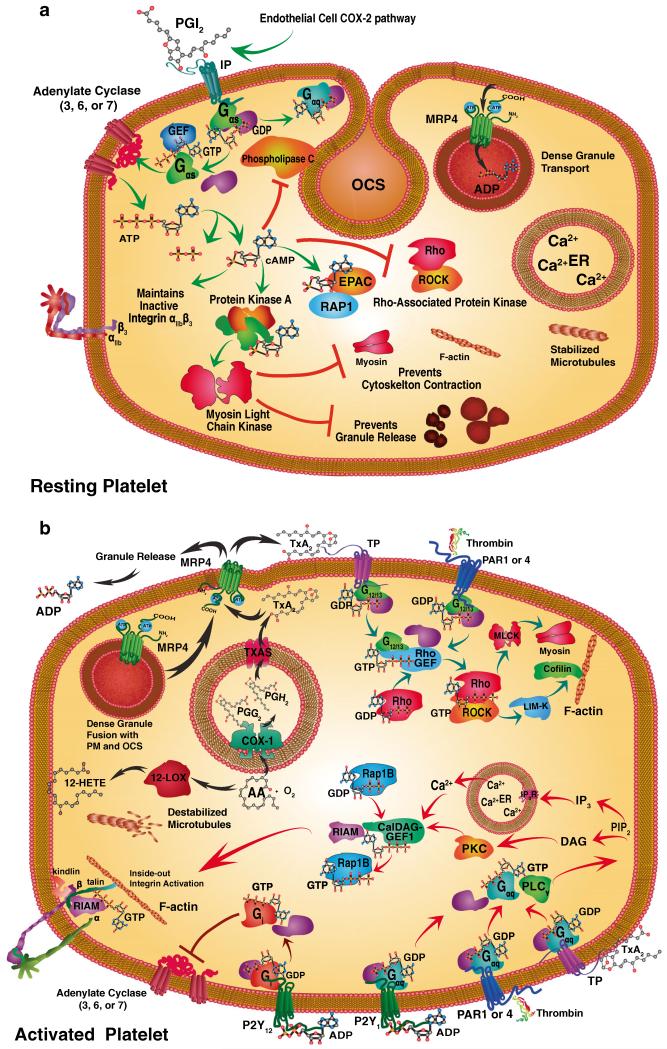
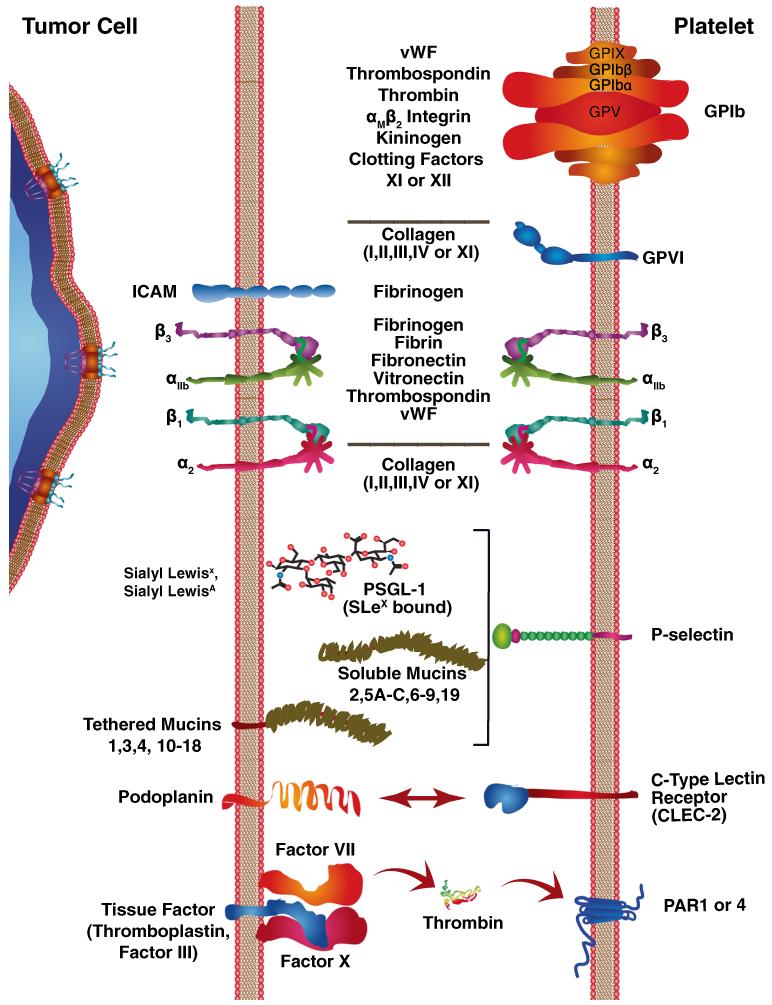
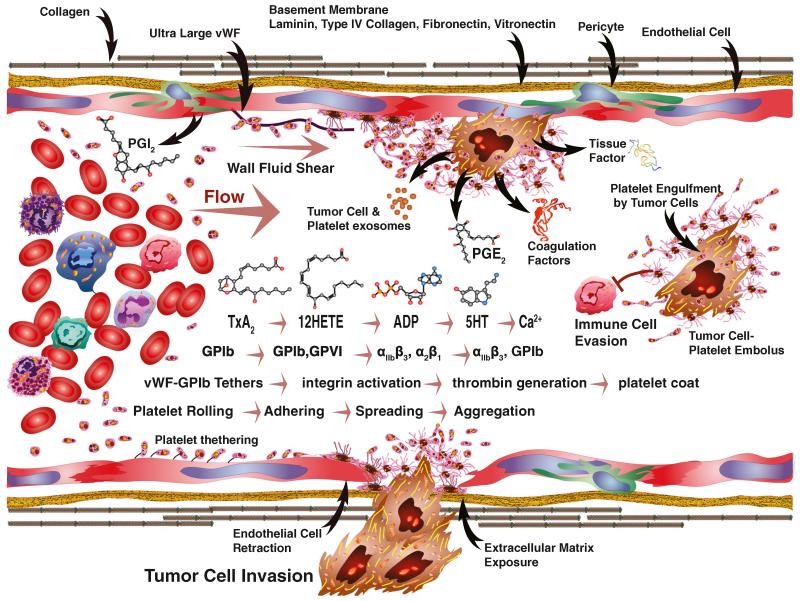
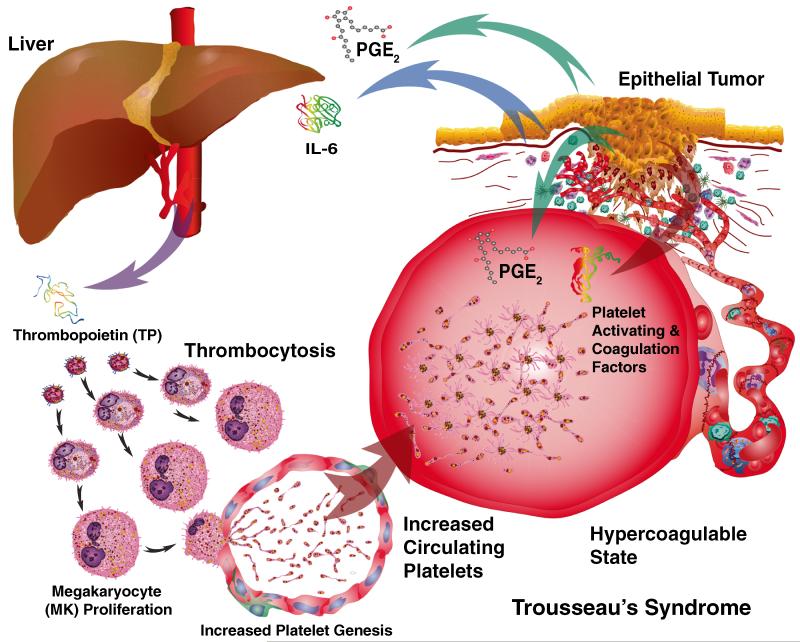
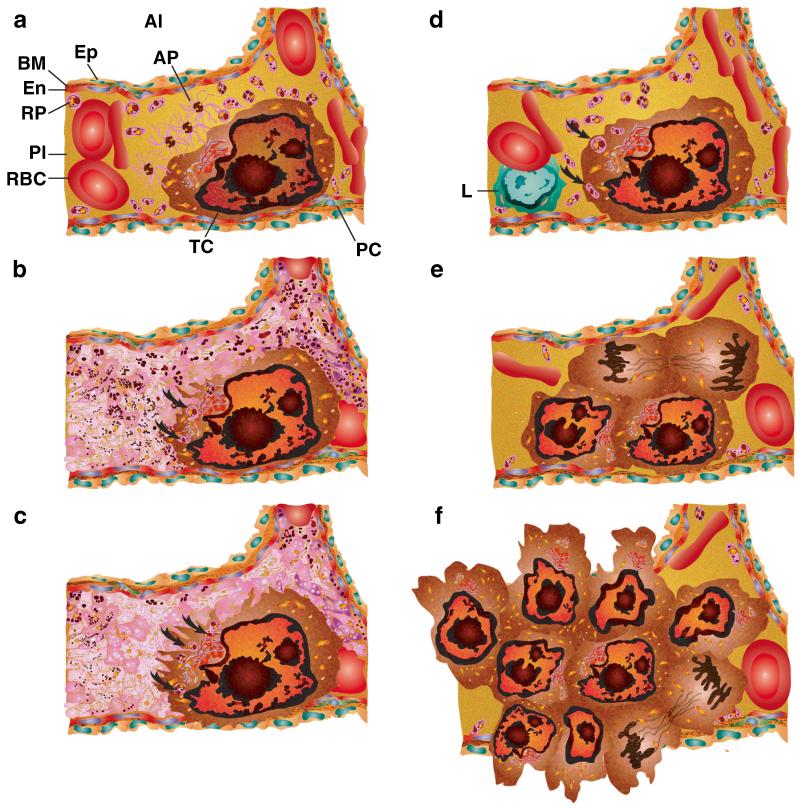
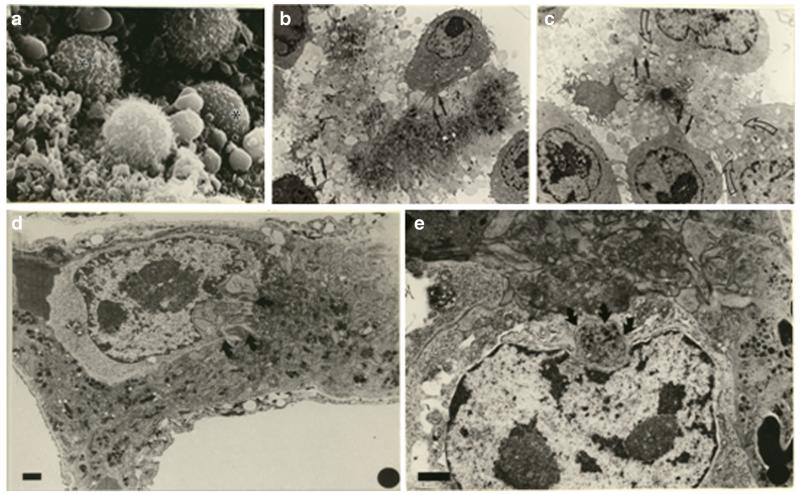
Human platelets arise as subcellular fragments of megakaryocytes in bone marrow. The physiologic demand, presence of disease such as cancer, or drug effects can regulate the production circulating platelets. Platelet biology is essential to hemostasis, vascular integrity, angiogenesis, inflammation, innate immunity, wound healing, and cancer biology. The most critical biological platelet response is serving as "First Responders" during the wounding process. The exposure of extracellular matrix proteins and intracellular components occurs after wounding. Numerous platelet receptors recognize matrix proteins that trigger platelet activation, adhesion, aggregation, and stabilization. Once activated, platelets change shape and degranulate to release growth factors and bioactive lipids into the blood stream. This cyclic process recruits and aggregates platelets along with thrombogenesis. This process facilitates wound closure or can recognize circulating pathologic bodies. Cancer cell entry into the blood stream triggers platelet-mediated recognition and is amplified by cell surface receptors, cellular products, extracellular factors, and immune cells. In some cases, these interactions suppress immune recognition and elimination of cancer cells or promote arrest at the endothelium, or entrapment in the microvasculature, and survival. This supports survival and spread of cancer cells and the establishment of secondary lesions to serve as important targets for prevention and therapy.
Figures
References
-
- Honn KV, Tang DG, Crissman JD. Platelets and cancer metastasis: a causal relationship? Cancer Metastasis Reviews. 1992;11(3-4):325–351. - PubMed
-
- Rados C. Beyond bloodletting: FDA gives leeches a medical makeover. FDA Consumer. 2004;38(5):9. - PubMed
-
- Winkel R, Tajsic N, Husum H, Schlageter M, Hanebuth G, Hoffmann R. Saphenous perforator flap. Operative Orthopädie und Traumatologie. 2013;25(2):152–161. - PubMed
-
- Steinhubl SR. Historical observations on the discovery of platelets, platelet function testing and the first antiplatelet agent. Current Drug Targets. 2011;12(12):1792–1804. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
