

The stress granule component TIA-1 binds tick-borne encephalitis virus RNA and is recruited to perinuclear sites of viral replication to inhibit viral translation
- PMID: 24696465
- PMCID: PMC4054376
- DOI: 10.1128/JVI.03736-13
The stress granule component TIA-1 binds tick-borne encephalitis virus RNA and is recruited to perinuclear sites of viral replication to inhibit viral translation
Abstract
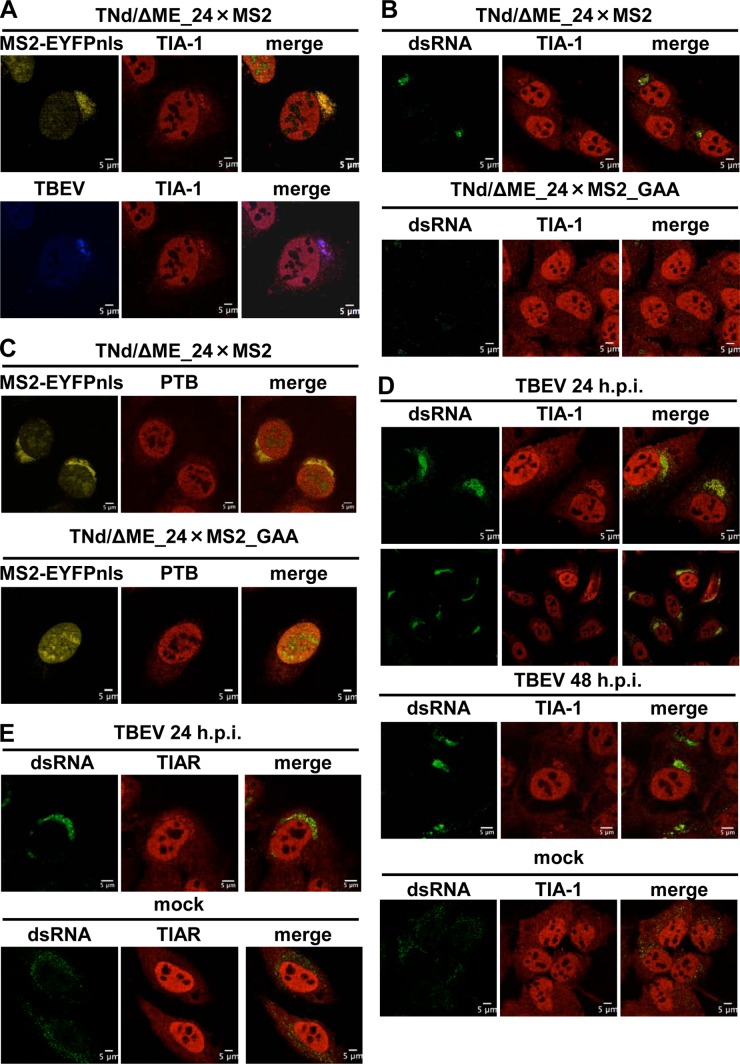
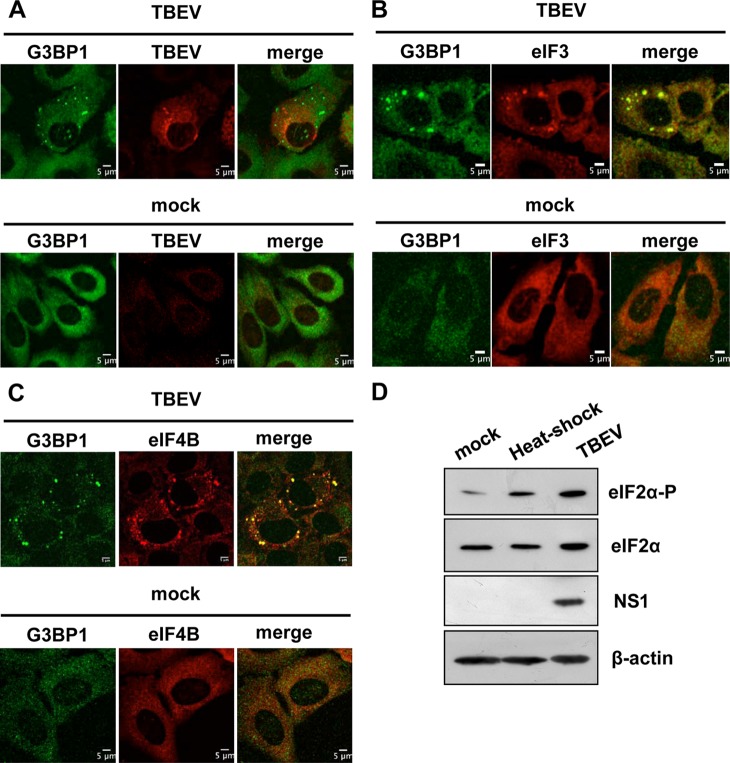
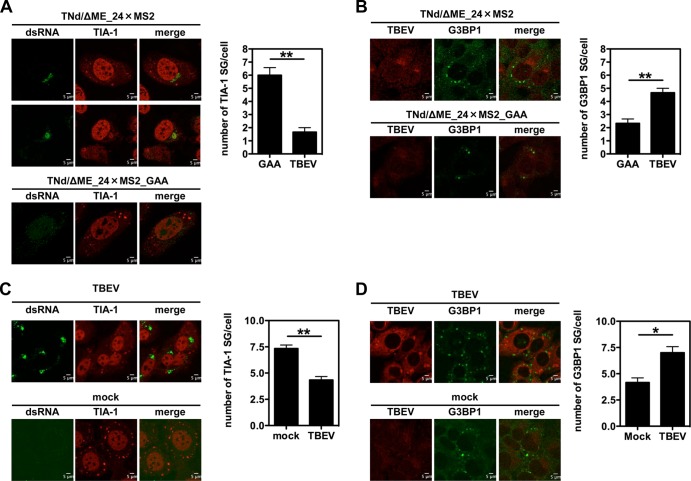
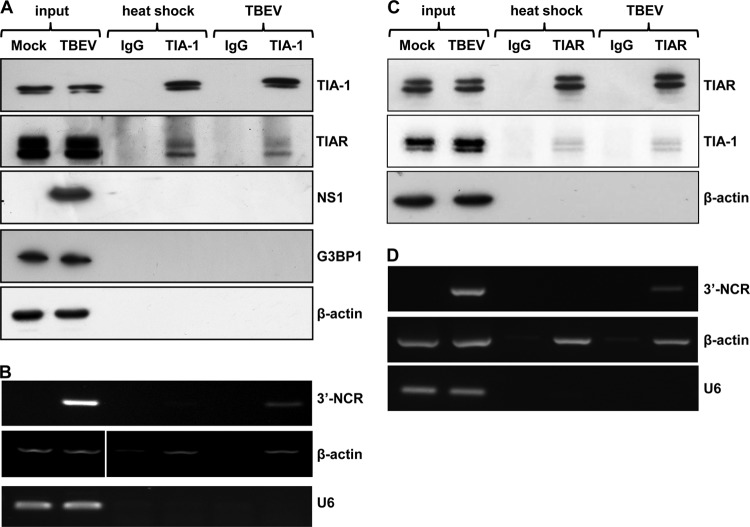
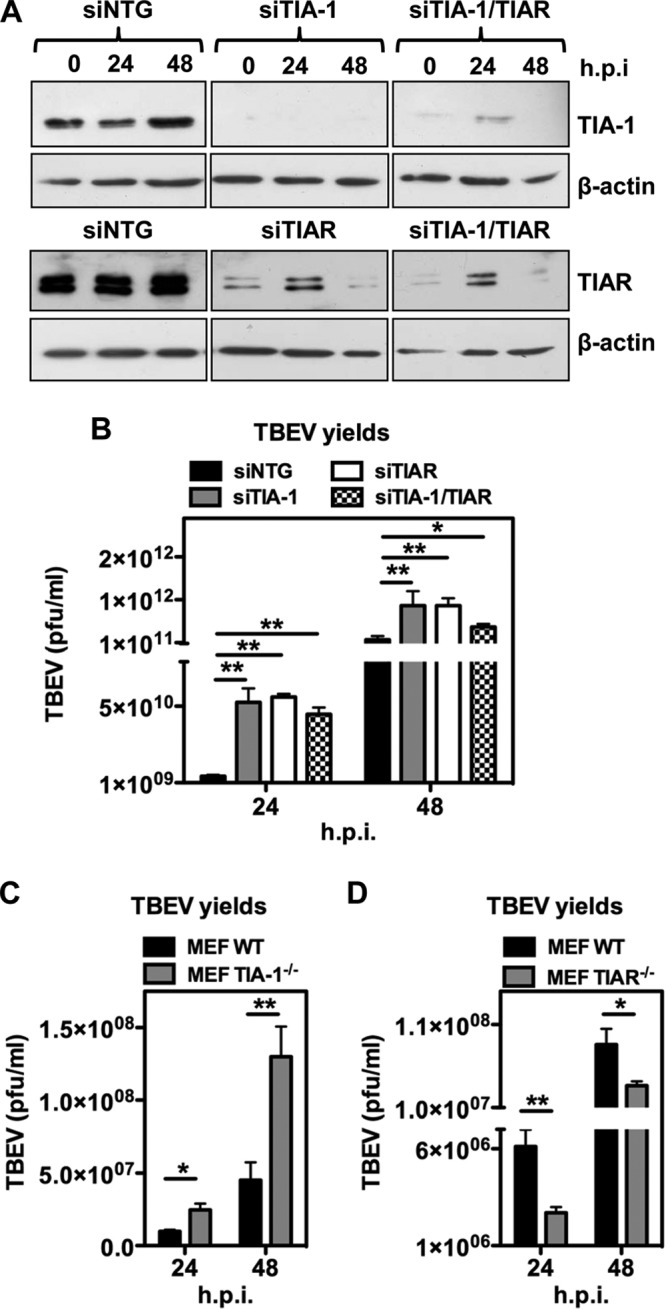
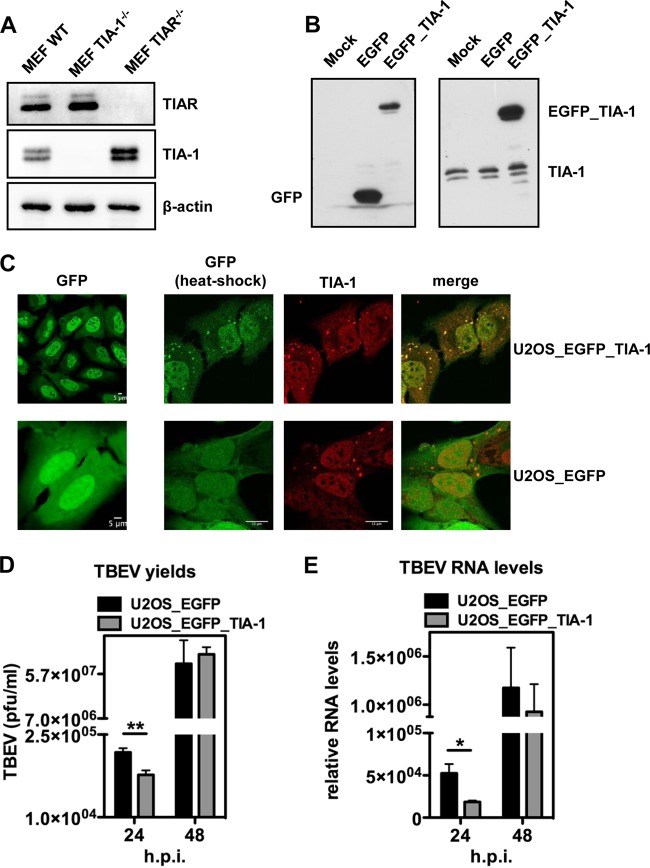
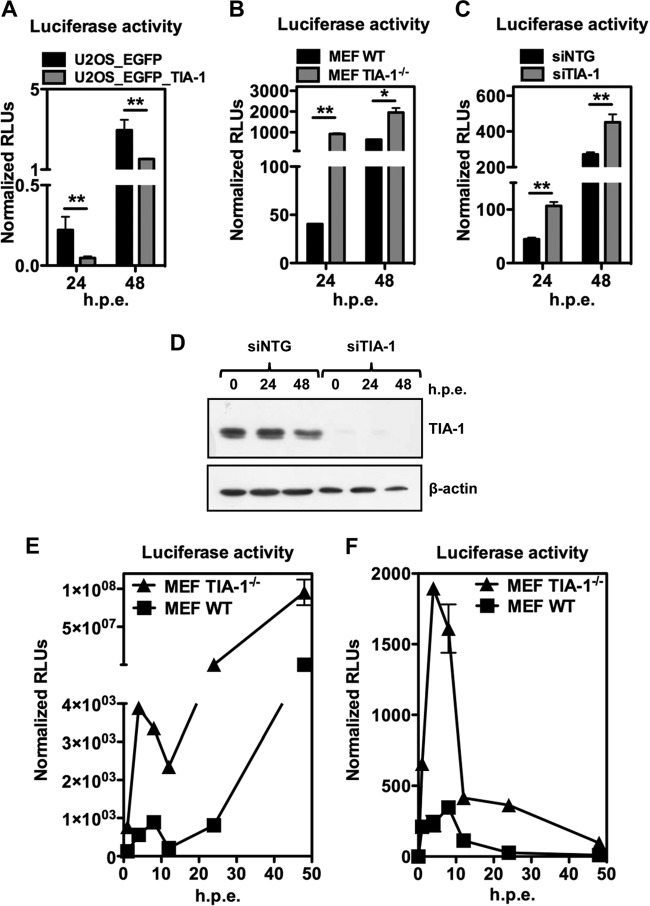
Flaviviruses are a major cause of disease in humans and animals worldwide. Tick-borne encephalitis virus (TBEV) is the most important arthropod-borne flavivirus endemic in Europe and is the etiological agent of tick-borne encephalitis, a potentially fatal infection of the central nervous system. However, the contributions of host proteins during TBEV infection are poorly understood. In this work, we investigate the cellular protein TIA-1 and its cognate factor TIAR, which are stress-induced RNA-binding proteins involved in the repression of initiation of translation of cellular mRNAs and in the formation of stress granules. We show that TIA-1 and TIAR interact with viral RNA in TBEV-infected cells. During TBEV infection, cytoplasmic TIA-1 and TIAR are recruited at sites of viral replication with concomitant depletion from stress granules. This effect is specific, since G3BP1, another component of these cytoplasmic structures, remains localized to stress granules. Moreover, heat shock induction of stress granules containing TIA-1, but not G3BP1, is inhibited in TBEV-infected cells. Infection of cells depleted of TIA-1 or TIAR by small interfering RNA (siRNA) or TIA-1(-/-) mouse fibroblasts, leads to a significant increase in TBEV extracellular infectivity. Interestingly, TIAR(-/-) fibroblasts show the opposite effect on TBEV infection, and this phenotype appears to be related to an excess of TIA-1 in these cells. Taking advantage of a TBE-luciferase replicon system, we also observed increased luciferase activity in TIA-1(-/-) mouse fibroblasts at early time points, consistent with TIA-1-mediated inhibition at the level of the first round of viral translation. These results indicate that, in response to TBEV infection, TIA-1 is recruited to sites of virus replication to bind TBEV RNA and modulate viral translation independently of stress granule (SG) formation.
Importance: This study (i) extends previous work that showed TIA-1/TIAR recruitment at sites of flavivirus replication, (ii) demonstrates that TIAR behaves like TIA-1 as an inhibitor of viral replication using an RNA interference (RNAi) approach in human cells that contradicts the previous hypothesis based on mouse embryonic fibroblast (MEF) knockouts only, (iii) demonstrates that tick-borne encephalitis virus (TBEV) is capable of inducing bona fide G3BP1/eIF3/eIF4B-positive stress granules, (iv) demonstrates a differential phenotype of stress response proteins following viral infection, and (v) implicates TIA-1 in viral translation and as a modulator of TBEV replication.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
References
-
- Lindenbach BD, Thiel HJ, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA. (ed), Fields virology, 5th ed. Lippincott, Williams & Wilkins, Philadelphia, PA
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
