

Confetti: a multiprotease map of the HeLa proteome for comprehensive proteomics
- PMID: 24696503
- PMCID: PMC4047476
- DOI: 10.1074/mcp.M113.035170
Confetti: a multiprotease map of the HeLa proteome for comprehensive proteomics
Abstract
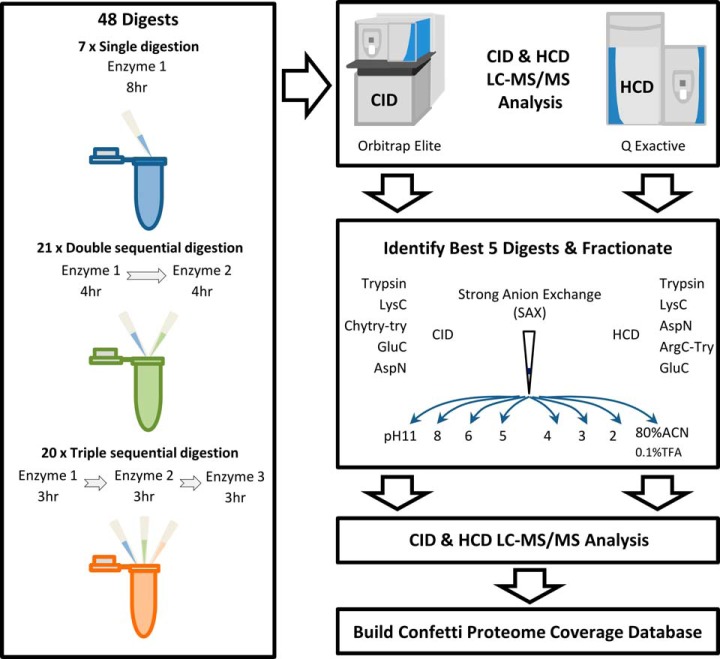
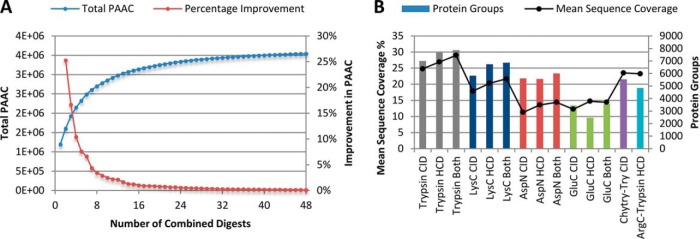
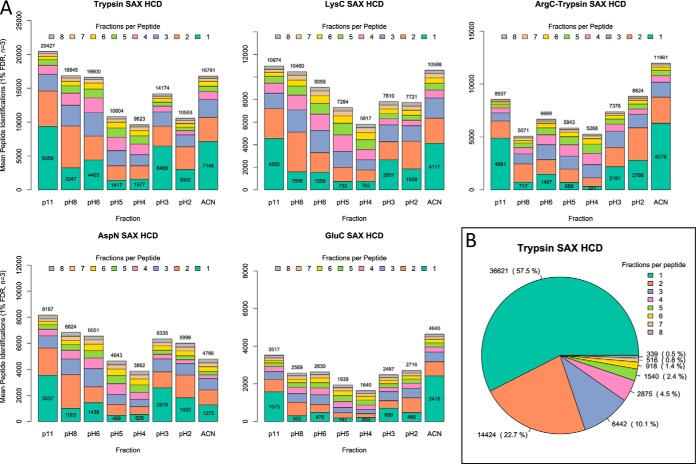
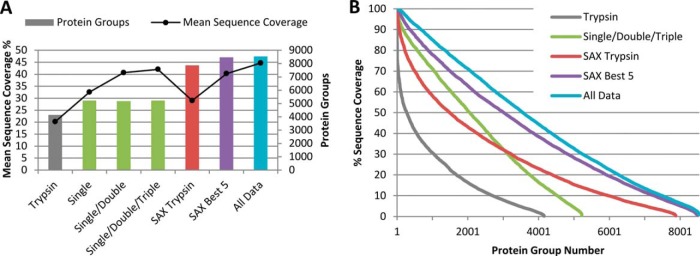
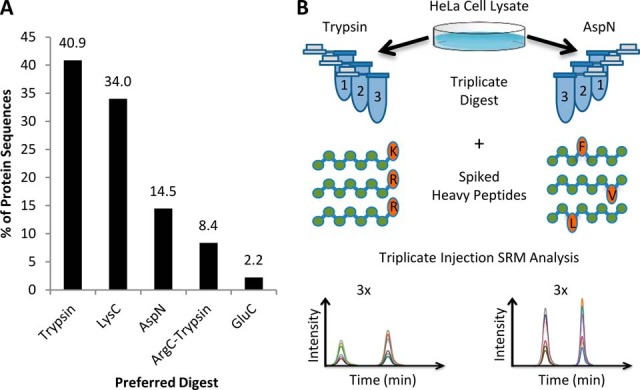
Bottom-up proteomics largely relies on tryptic peptides for protein identification and quantification. Tryptic digestion often provides limited coverage of protein sequence because of issues such as peptide length, ionization efficiency, and post-translational modification colocalization. Unfortunately, a region of interest in a protein, for example, because of proximity to an active site or the presence of important post-translational modifications, may not be covered by tryptic peptides. Detection limits, quantification accuracy, and isoform differentiation can also be improved with greater sequence coverage. Selected reaction monitoring (SRM) would also greatly benefit from being able to identify additional targetable sequences. In an attempt to improve protein sequence coverage and to target regions of proteins that do not generate useful tryptic peptides, we deployed a multiprotease strategy on the HeLa proteome. First, we used seven commercially available enzymes in single, double, and triple enzyme combinations. A total of 48 digests were performed. 5223 proteins were detected by analyzing the unfractionated cell lysate digest directly; with 42% mean sequence coverage. Additional strong-anion exchange fractionation of the most complementary digests permitted identification of over 3000 more proteins, with improved mean sequence coverage. We then constructed a web application (https://proteomics.swmed.edu/confetti) that allows the community to examine a target protein or protein isoform in order to discover the enzyme or combination of enzymes that would yield peptides spanning a certain region of interest in the sequence. Finally, we examined the use of nontryptic digests for SRM. From our strong-anion exchange fractionation data, we were able to identify three or more proteotypic SRM candidates within a single digest for 6056 genes. Surprisingly, in 25% of these cases the digest producing the most observable proteotypic peptides was neither trypsin nor Lys-C. SRM analysis of Asp-N versus tryptic peptides for eight proteins determined that Asp-N yielded higher signal in five of eight cases.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures

References
-
- Eichacker L. A., Granvogl B., Mirus O., Muller B. C., Miess C., Schleiff E. (2004) Hiding behind hydrophobicity: transmembrane segments in mass spectrometry. J. Biol. Chem. 279, 50915–50922 - PubMed
-
- Meyer B., Papasotiriou D. G., Karas M. (2011) 100% protein sequence coverage: a modern form of surrealism in proteomics. Amino Acids 41, 291–310 - PubMed
-
- Nature Methods (2013) Method of the Year 2012. Nat. Methods 10, 1. - PubMed
-
- Ebhardt H. A., Sabido E., Huttenhain R., Collins B., Aebersold R. (2012) Range of protein detection by selected/multiple reaction monitoring mass spectrometry in an unfractionated human cell culture lysate. Proteomics 12, 1185–1193 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
