

Cantú syndrome resulting from activating mutation in the KCNJ8 gene
- PMID: 24700710
- PMCID: PMC4277879
- DOI: 10.1002/humu.22555
Cantú syndrome resulting from activating mutation in the KCNJ8 gene
Abstract
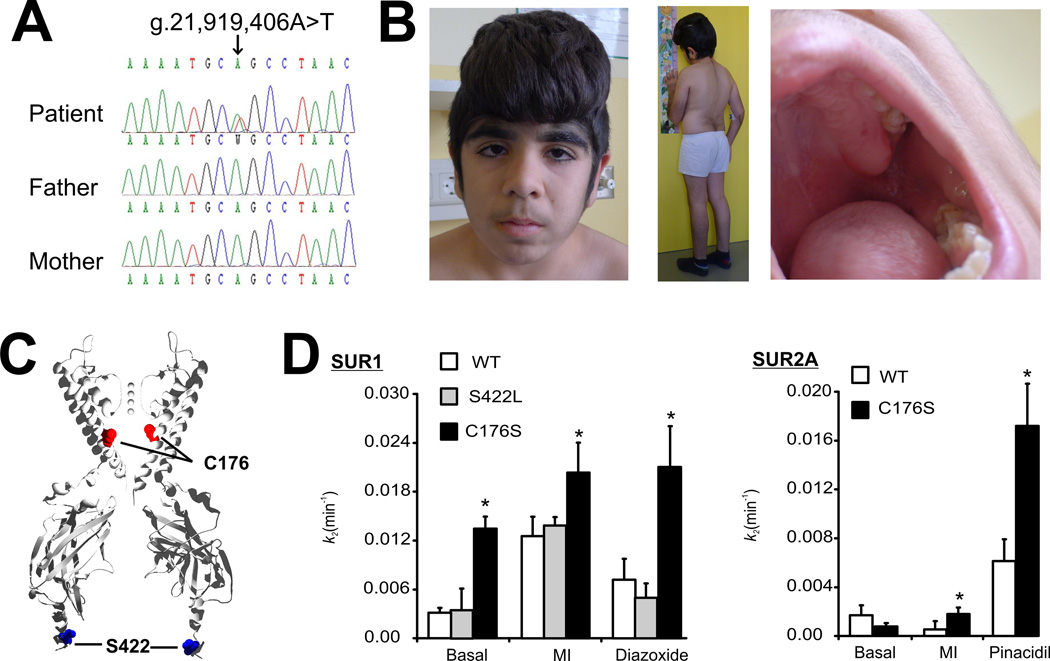
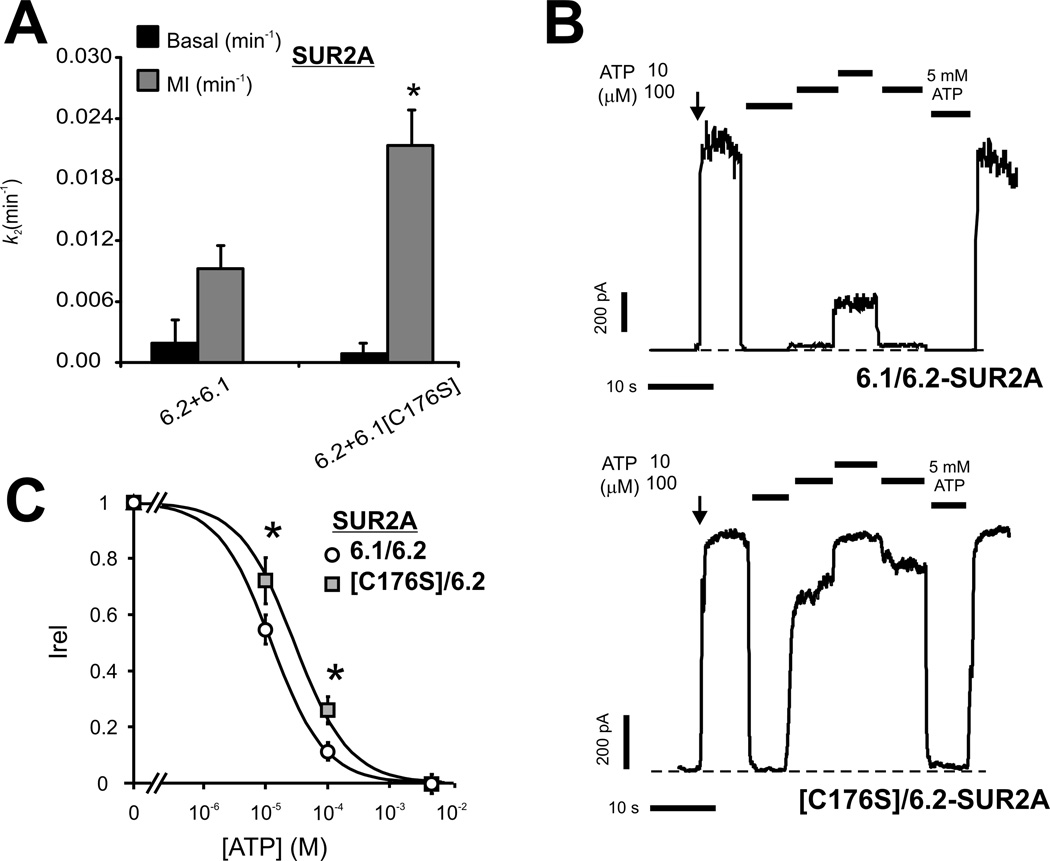
ATP-sensitive potassium (KATP ) channels, composed of inward-rectifying potassium channel subunits (Kir6.1 and Kir6.2, encoded by KCNJ8 and KCNJ11, respectively) and regulatory sulfonylurea receptor (SUR1 and SUR2, encoded by ABCC8 and ABCC9, respectively), couple metabolism to excitability in multiple tissues. Mutations in ABCC9 cause Cantú syndrome (CS), a distinct multiorgan disease, potentially via enhanced KATP channel activity. We screened KCNJ8 in an ABCC9 mutation-negative patient who also exhibited clinical hallmarks of CS (hypertrichosis, macrosomia, macrocephaly, coarse facial appearance, cardiomegaly, and skeletal abnormalities). We identified a de novo missense mutation encoding Kir6.1[p.Cys176Ser] in the patient. Kir6.1[p.Cys176Ser] channels exhibited markedly higher activity than wild-type channels, as a result of reduced ATP sensitivity, whether coexpressed with SUR1 or SUR2A subunits. Our results identify a novel causal gene in CS, but also demonstrate that the cardinal features of the disease result from gain of KATP channel function, not from a Kir6-independent SUR2 function.
Keywords: Cantú syndrome; KATP; KCNJ8; Kir6.1; hypertrichosis.
© 2014 WILEY PERIODICALS, INC.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Brownstein CA, Towne MC, Luquette LJ, Harris DJ, Marinakis NS, Meinecke P, Kutsche K, Campeau PM, Yu TW, Margulies DM, et al. Mutation of KCNJ8 in a patient with Cantu syndrome with unique vascular abnormalities - Support for the role of K(ATP) channels in this condition. Eur J Med Genet. 2013;56:678–682. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
