

Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families
- PMID: 24702956
- PMCID: PMC3980410
- DOI: 10.1016/j.ajhg.2014.03.010
Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families
Abstract
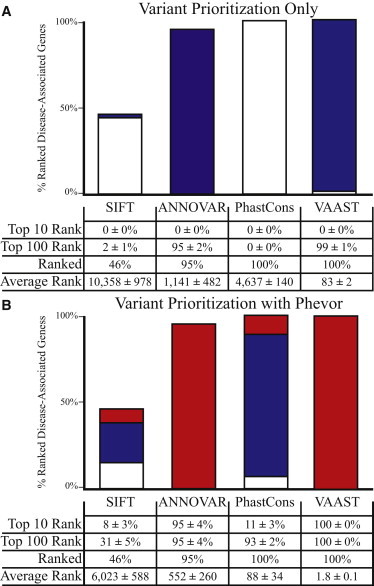
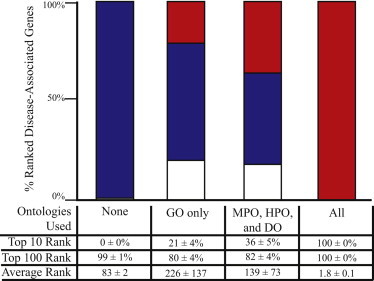
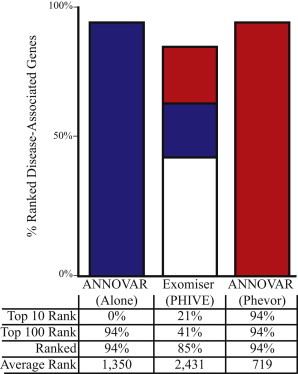
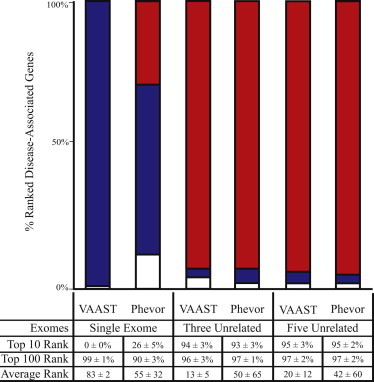
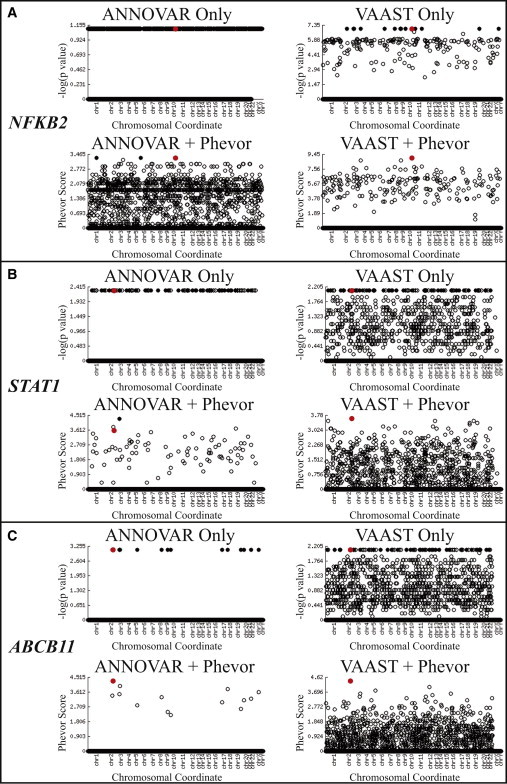
Phevor integrates phenotype, gene function, and disease information with personal genomic data for improved power to identify disease-causing alleles. Phevor works by combining knowledge resident in multiple biomedical ontologies with the outputs of variant-prioritization tools. It does so by using an algorithm that propagates information across and between ontologies. This process enables Phevor to accurately reprioritize potentially damaging alleles identified by variant-prioritization tools in light of gene function, disease, and phenotype knowledge. Phevor is especially useful for single-exome and family-trio-based diagnostic analyses, the most commonly occurring clinical scenarios and ones for which existing personal genome diagnostic tools are most inaccurate and underpowered. Here, we present a series of benchmark analyses illustrating Phevor's performance characteristics. Also presented are three recent Utah Genome Project case studies in which Phevor was used to identify disease-causing alleles. Collectively, these results show that Phevor improves diagnostic accuracy not only for individuals presenting with established disease phenotypes but also for those with previously undescribed and atypical disease presentations. Importantly, Phevor is not limited to known diseases or known disease-causing alleles. As we demonstrate, Phevor can also use latent information in ontologies to discover genes and disease-causing alleles not previously associated with disease.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Chen K., Coonrod E.M., Kumánovics A., Franks Z.F., Durtschi J.D., Margraf R.L., Wu W., Heikal N.M., Augustine N.H., Ridge P.G. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 2013;93:812–824. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
