

The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma
- PMID: 24705251
- PMCID: PMC4056452
- DOI: 10.1038/ng.2938
The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma
Abstract
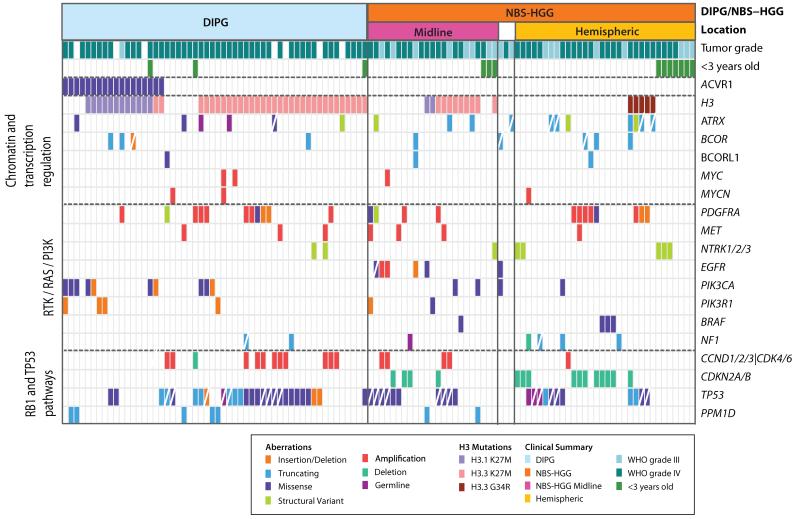
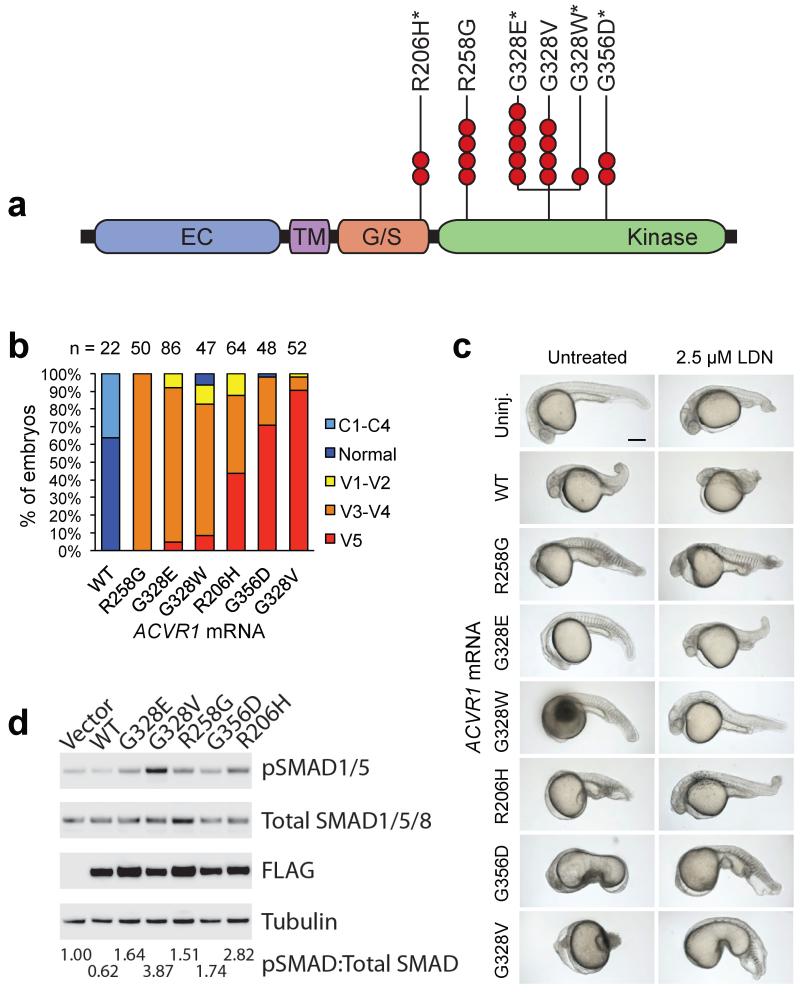
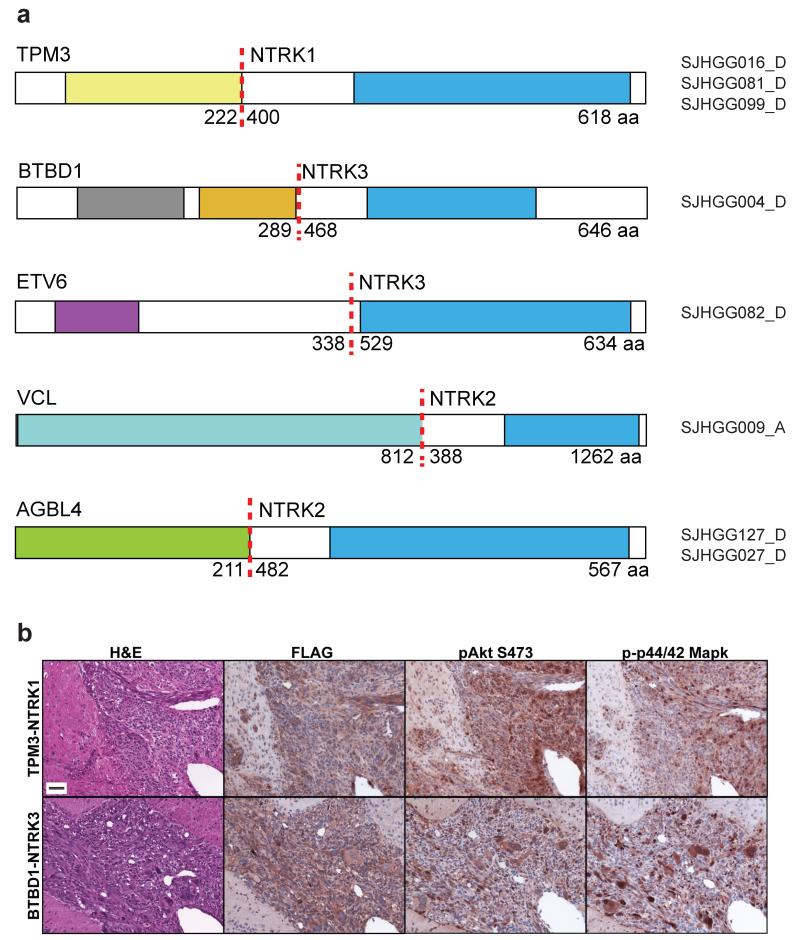
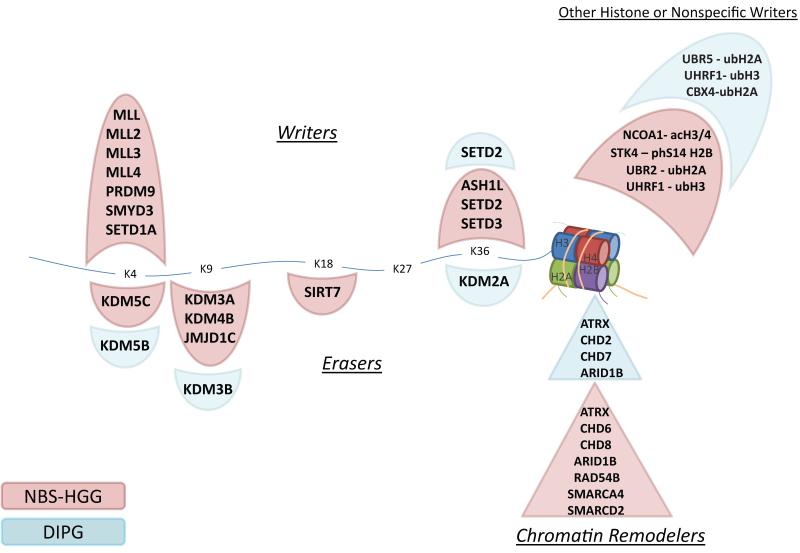
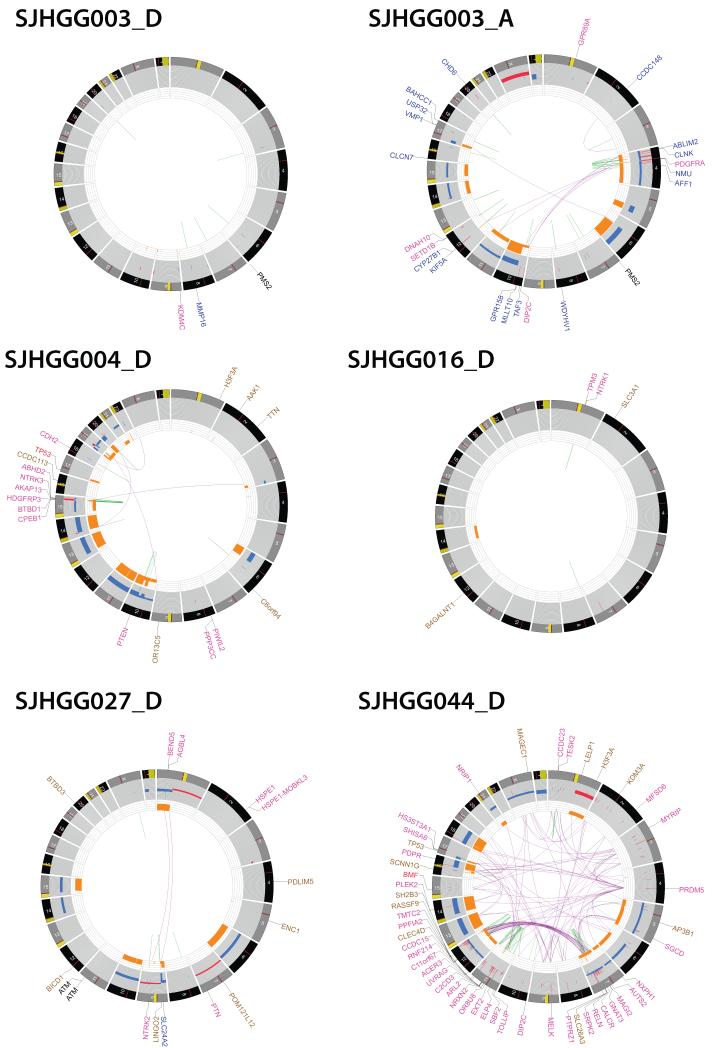
Pediatric high-grade glioma (HGG) is a devastating disease with a less than 20% survival rate 2 years after diagnosis. We analyzed 127 pediatric HGGs, including diffuse intrinsic pontine gliomas (DIPGs) and non-brainstem HGGs (NBS-HGGs), by whole-genome, whole-exome and/or transcriptome sequencing. We identified recurrent somatic mutations in ACVR1 exclusively in DIPGs (32%), in addition to previously reported frequent somatic mutations in histone H3 genes, TP53 and ATRX, in both DIPGs and NBS-HGGs. Structural variants generating fusion genes were found in 47% of DIPGs and NBS-HGGs, with recurrent fusions involving the neurotrophin receptor genes NTRK1, NTRK2 and NTRK3 in 40% of NBS-HGGs in infants. Mutations targeting receptor tyrosine kinase-RAS-PI3K signaling, histone modification or chromatin remodeling, and cell cycle regulation were found in 68%, 73% and 59% of pediatric HGGs, respectively, including in DIPGs and NBS-HGGs. This comprehensive analysis provides insights into the unique and shared pathways driving pediatric HGG within and outside the brainstem.
Figures
Comment in
-
ACVR1 mutations and the genomic landscape of pediatric diffuse glioma.Nat Genet. 2014 May;46(5):421-2. doi: 10.1038/ng.2970. Nat Genet. 2014. PMID: 24769718 No abstract available.
-
Genetics: ACVR1 mutations-a key piece in paediatric diffuse glioma.Nat Rev Clin Oncol. 2014 Jun;11(6):300. doi: 10.1038/nrclinonc.2014.74. Epub 2014 May 20. Nat Rev Clin Oncol. 2014. PMID: 24840074 No abstract available.
References
-
- Pollack IF, et al. Age and TP53 mutation frequency in childhood malignant gliomas: results in a multi-institutional cohort. Cancer Res. 2001;61:7404–7. - PubMed
-
- Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
