

Single-molecule spectroscopy of amino acids and peptides by recognition tunnelling
- PMID: 24705512
- PMCID: PMC4047173
- DOI: 10.1038/nnano.2014.54
Single-molecule spectroscopy of amino acids and peptides by recognition tunnelling
Abstract
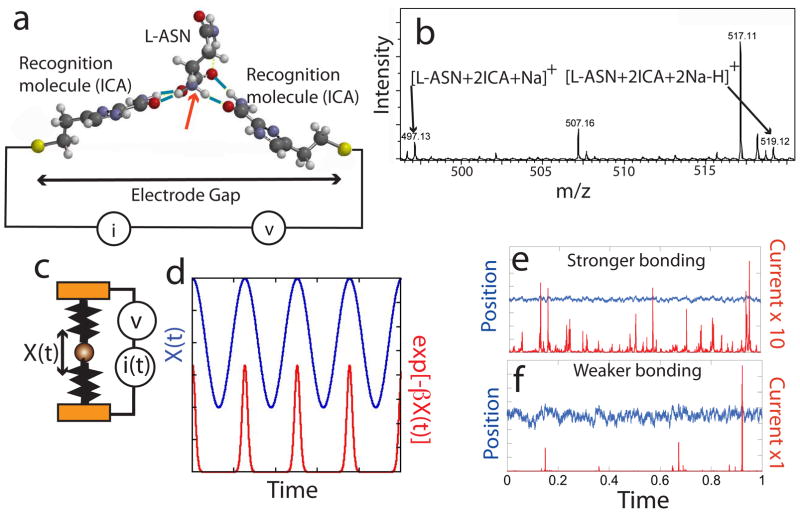
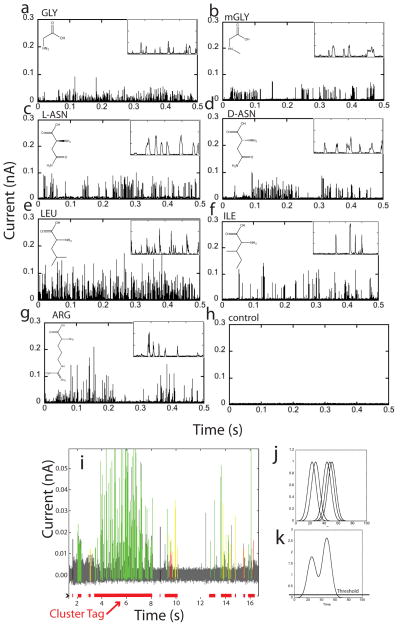
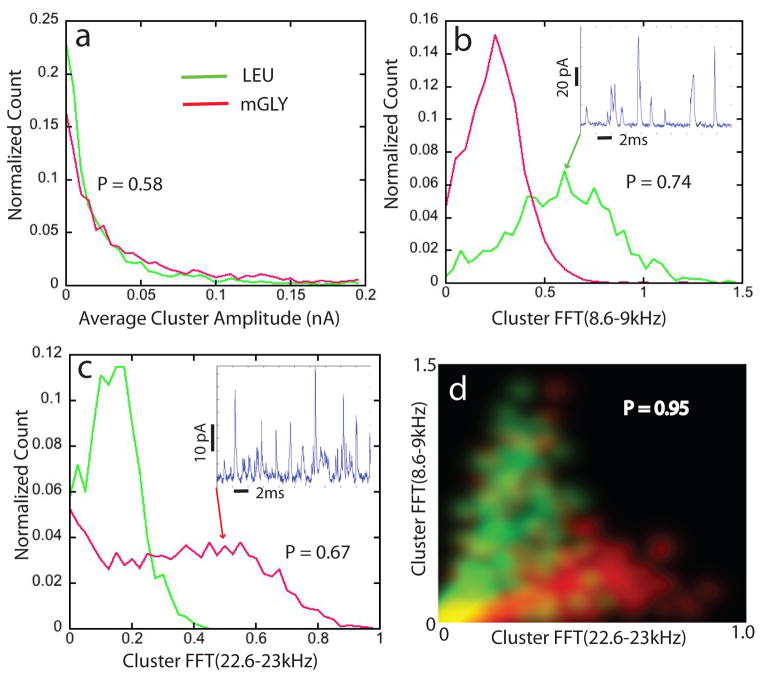
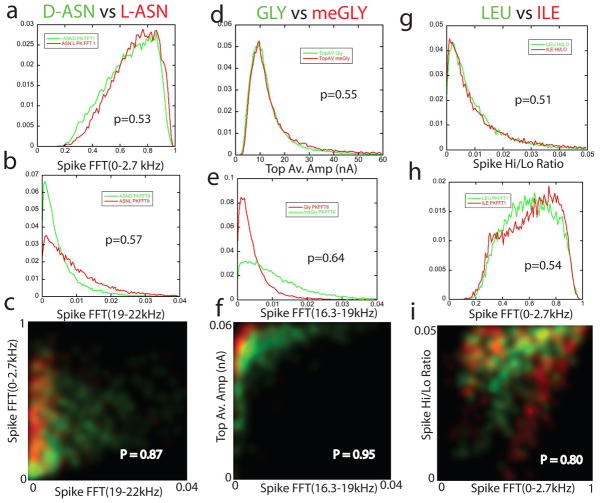
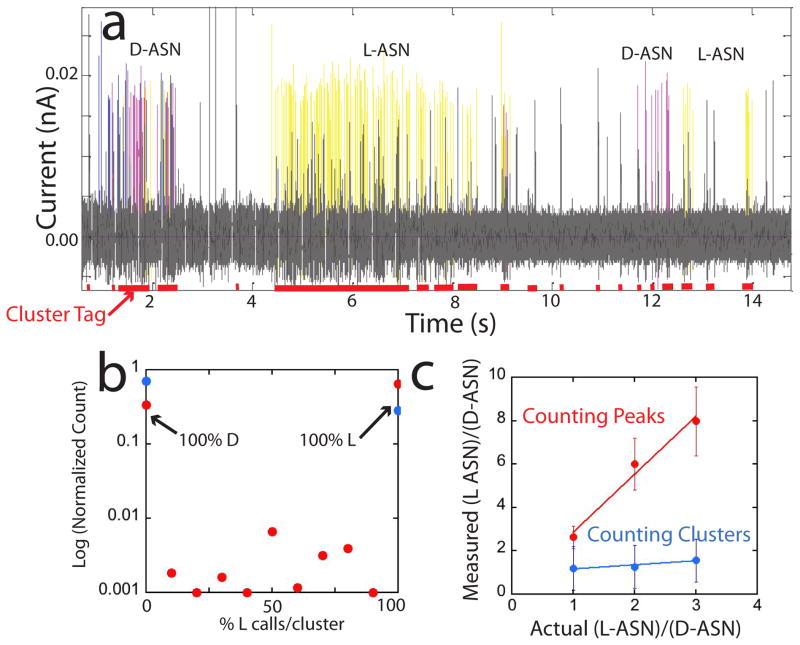
The human proteome has millions of protein variants due to alternative RNA splicing and post-translational modifications, and variants that are related to diseases are frequently present in minute concentrations. For DNA and RNA, low concentrations can be amplified using the polymerase chain reaction, but there is no such reaction for proteins. Therefore, the development of single-molecule protein sequencing is a critical step in the search for protein biomarkers. Here, we show that single amino acids can be identified by trapping the molecules between two electrodes that are coated with a layer of recognition molecules, then measuring the electron tunnelling current across the junction. A given molecule can bind in more than one way in the junction, and we therefore use a machine-learning algorithm to distinguish between the sets of electronic 'fingerprints' associated with each binding motif. With this recognition tunnelling technique, we are able to identify D and L enantiomers, a methylated amino acid, isobaric isomers and short peptides. The results suggest that direct electronic sequencing of single proteins could be possible by sequentially measuring the products of processive exopeptidase digestion, or by using a molecular motor to pull proteins through a tunnel junction integrated with a nanopore.
Conflict of interest statement
YZ, PZ and SL are named as inventors in patent applications. SL is cofounder of a company based on this technology.
Figures
References
-
- Uhlen M, Ponten F. Antibody-based Proteomics for Human Tissue Profiling. Molecular & Cellular Proteomics. 2005;4:384–393. - PubMed
-
- National Research Council (US) Committee on Intellectual Property Rights in Genomic and Protein Research and Innovation. Reaping the Benefits of Genomic and Proteomic Research: Intellectual Property Rights, Innovation, and Public Health. National Academies Press (US); 2006. - PubMed
-
- Archakov AI, Ivanov YD, Lisitsa AV, Zgoda VG. AFM fishing nanotechnology is the way to reverse the Avogadro number in proteomics. Proteomics. 2007;7:4–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
