

Distinct phenotype of a Wilson disease mutation reveals a novel trafficking determinant in the copper transporter ATP7B
- PMID: 24706876
- PMCID: PMC3986166
- DOI: 10.1073/pnas.1314161111
Distinct phenotype of a Wilson disease mutation reveals a novel trafficking determinant in the copper transporter ATP7B
Abstract
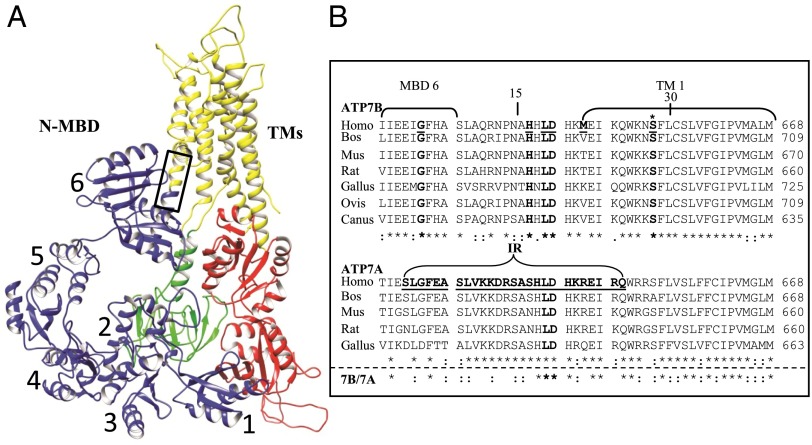
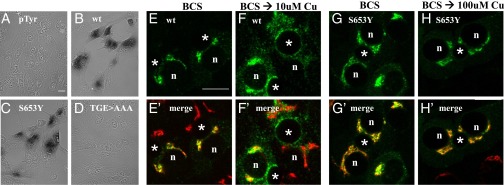
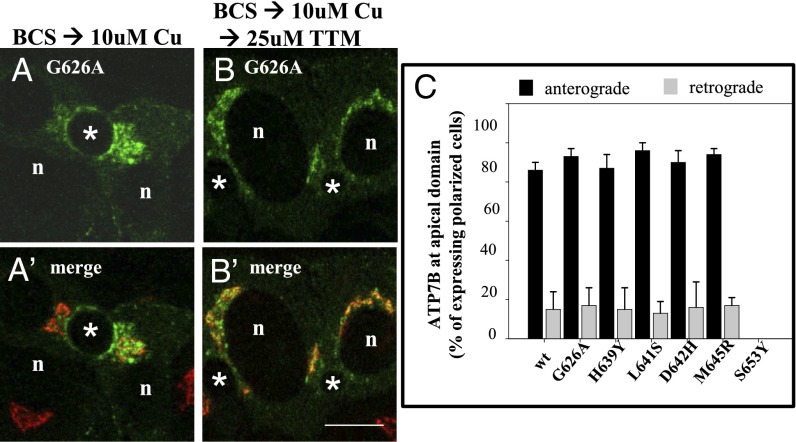
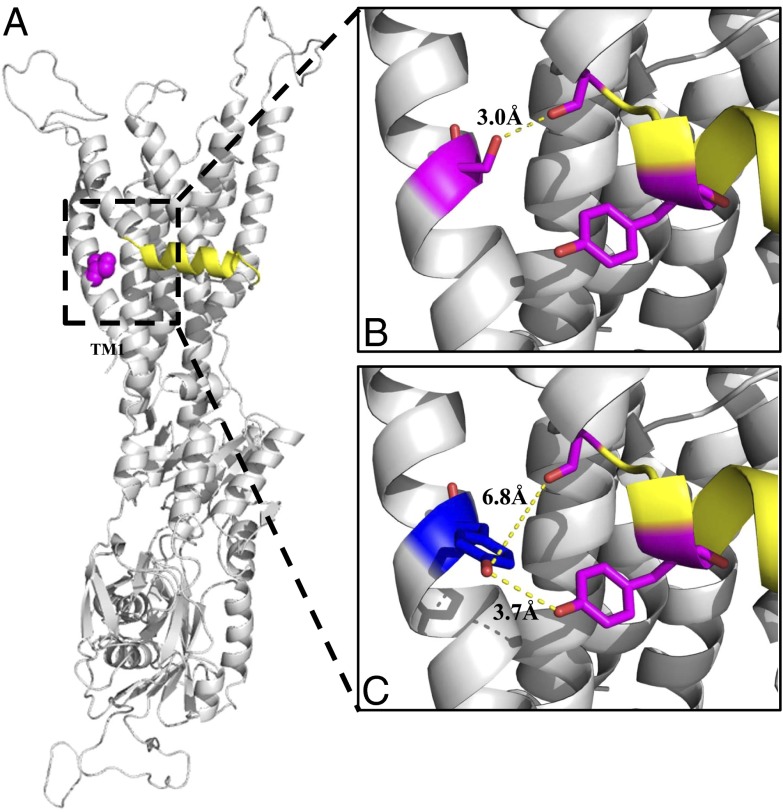
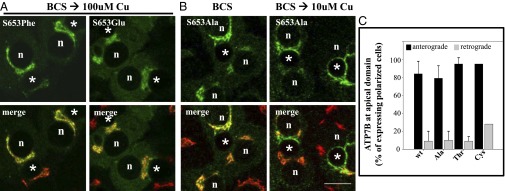
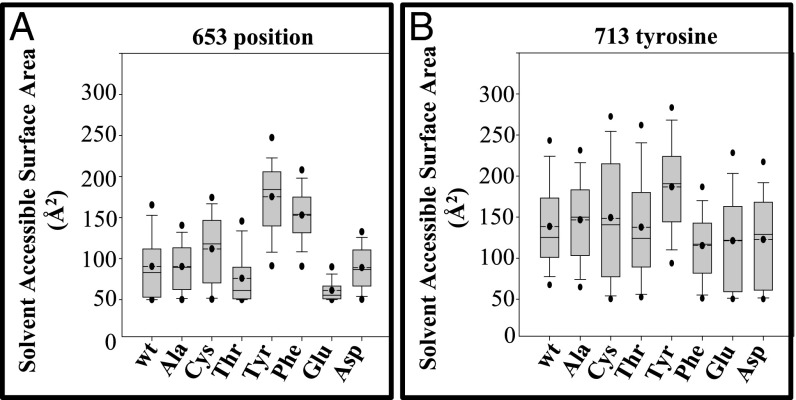
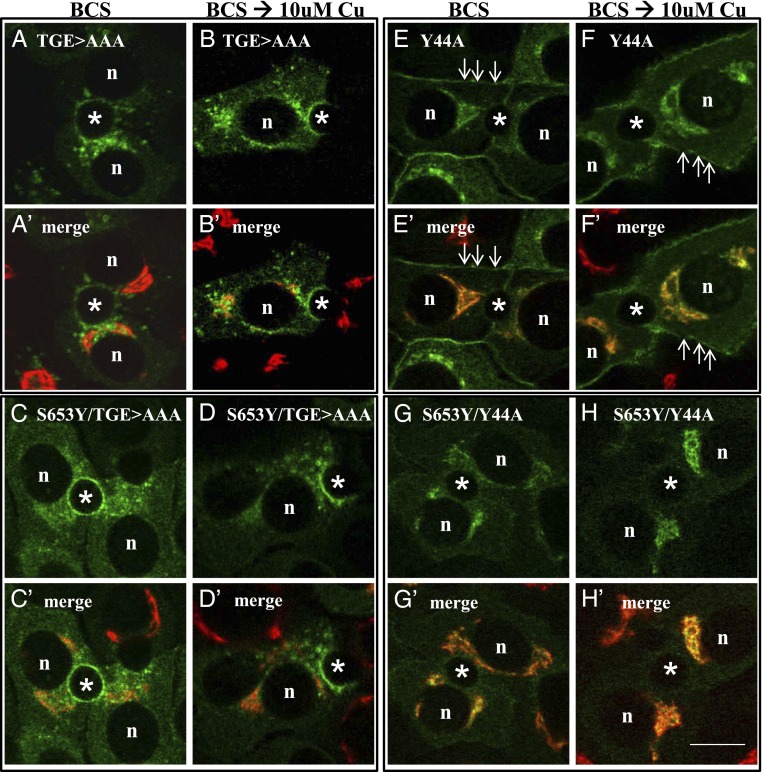
Wilson disease (WD) is a monogenic autosomal-recessive disorder of copper accumulation that leads to liver failure and/or neurological deficits. WD is caused by mutations in ATP7B, a transporter that loads Cu(I) onto newly synthesized cupro-enzymes in the trans-Golgi network (TGN) and exports excess copper out of cells by trafficking from the TGN to the plasma membrane. To date, most WD mutations have been shown to disrupt ATP7B activity and/or stability. Using a multidisciplinary approach, including clinical analysis of patients, cell-based assays, and computational studies, we characterized a patient mutation, ATP7B(S653Y), which is stable, does not disrupt Cu(I) transport, yet renders the protein unable to exit the TGN. Bulky or charged substitutions at position 653 mimic the phenotype of the patient mutation. Molecular modeling and dynamic simulation suggest that the S653Y mutation induces local distortions within the transmembrane (TM) domain 1 and alter TM1 interaction with TM2. S653Y abolishes the trafficking-stimulating effects of a secondary mutation in the N-terminal apical targeting domain. This result indicates a role for TM1/TM2 in regulating conformations of cytosolic domains involved in ATP7B trafficking. Taken together, our experiments revealed an unexpected role for TM1/TM2 in copper-regulated trafficking of ATP7B and defined a unique class of WD mutants that are transport-competent but trafficking-defective. Understanding the precise consequences of WD-causing mutations will facilitate the development of advanced mutation-specific therapies.
Keywords: ceruloplasmin; interdomain interactions; molecular dynamics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87(3):1011–1046. - PubMed
-
- Ferenci P, Roberts EA. Defining Wilson disease phenotypes: From the patient to the bench and back again. Gastroenterology. 2012;142(4):692–696. - PubMed
-
- Forbes JR, Cox DW. Copper-dependent trafficking of Wilson disease mutant ATP7B proteins. Hum Mol Genet. 2000;9(13):1927–1935. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
