

The docking protein FRS2α is a critical regulator of VEGF receptors signaling
- PMID: 24706887
- PMCID: PMC3992672
- DOI: 10.1073/pnas.1404545111
The docking protein FRS2α is a critical regulator of VEGF receptors signaling
Abstract
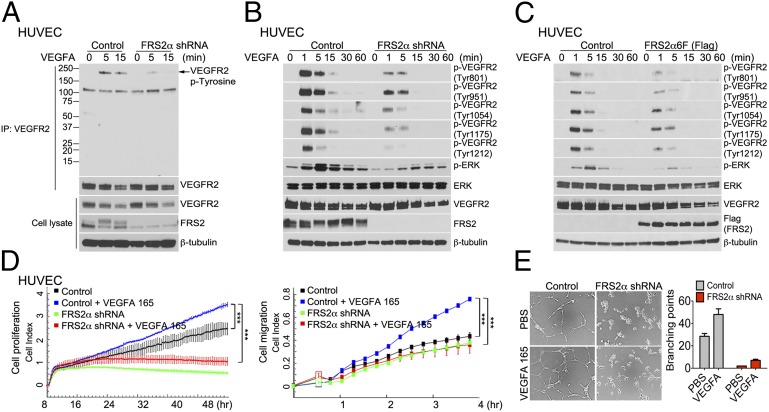
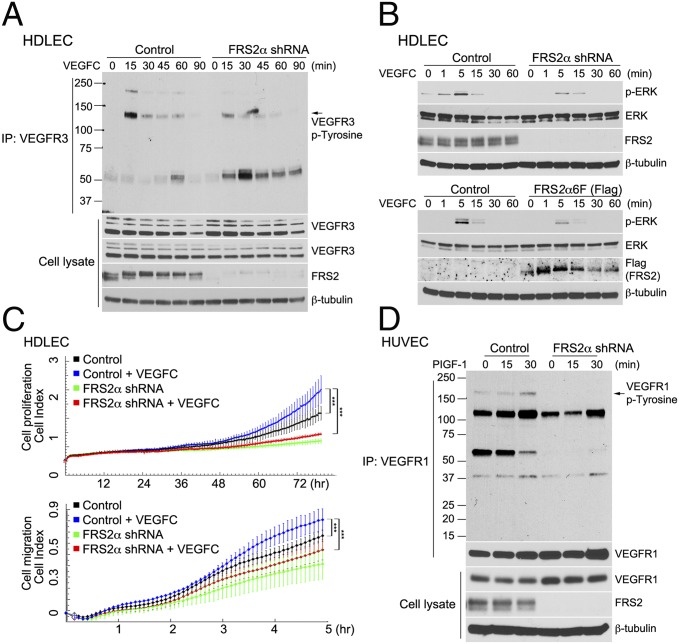
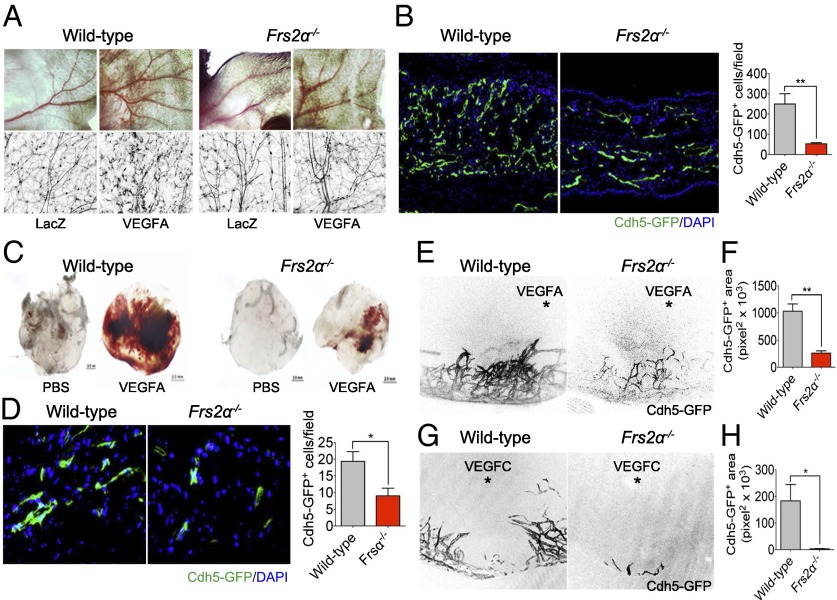
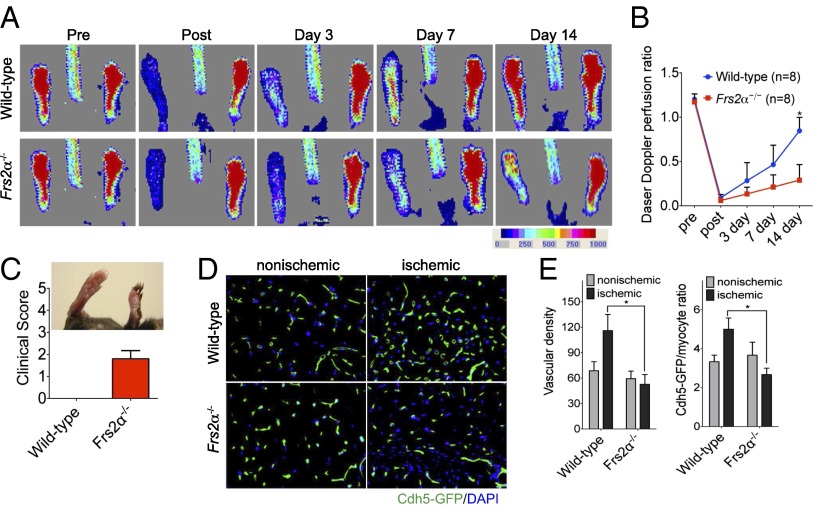
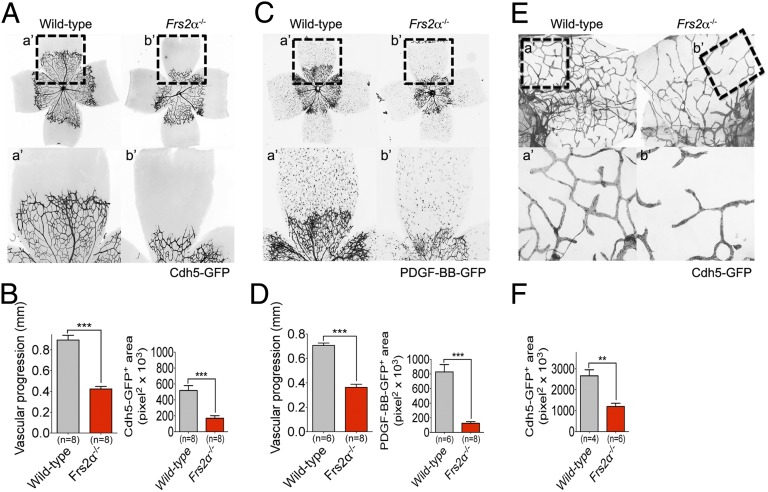
Vascular endothelial growth factors (VEGFs) signal via their cognate receptor tyrosine kinases designated VEGFR1-3. We report that the docking protein fibroblast growth factor receptor substrate 2 (FRS2α) plays a critical role in cell signaling via these receptors. In vitro FRS2α regulates VEGF-A and VEGF-C-dependent activation of extracellular signal-regulated receptor kinase signaling and blood and lymphatic endothelial cells migration and proliferation. In vivo endothelial-specific deletion of FRS2α results in the profound impairment of postnatal vascular development and adult angiogenesis, lymphangiogenesis, and arteriogenesis. We conclude that FRS2α is a previously unidentified component of VEGF receptors signaling.
Keywords: FGF receptor; MAP kinase; phosphorylation; receptor kinase inhibition; signal transduction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21(2):154–165. - PubMed
-
- Benedito R, et al. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484(7392):110–114. - PubMed
-
- Wang Y, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465(7297):483–486. - PubMed
-
- Sawamiphak S, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465(7297):487–491. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
