

Slit2N and Robo4 regulate lymphangiogenesis through the VEGF-C/VEGFR-3 pathway
- PMID: 24708522
- PMCID: PMC4122147
- DOI: 10.1186/1478-811X-12-25
Slit2N and Robo4 regulate lymphangiogenesis through the VEGF-C/VEGFR-3 pathway
Abstract
Background: Signaling through vascular endothelial growth factor C (VEGF–C) and VEGF receptor 3 (VEGFR-3) plays a central role in lymphangiogenesis and the metastasis of several cancers via the lymphatics. Recently, the Slit2/Robo4 pathway has been recognized as a modulator of vascular permeability and integrity. Signaling via the Robo receptor inhibits VEGF-mediated effects; however, its effects on lymphatic endothelial cell function have not been well characterized.
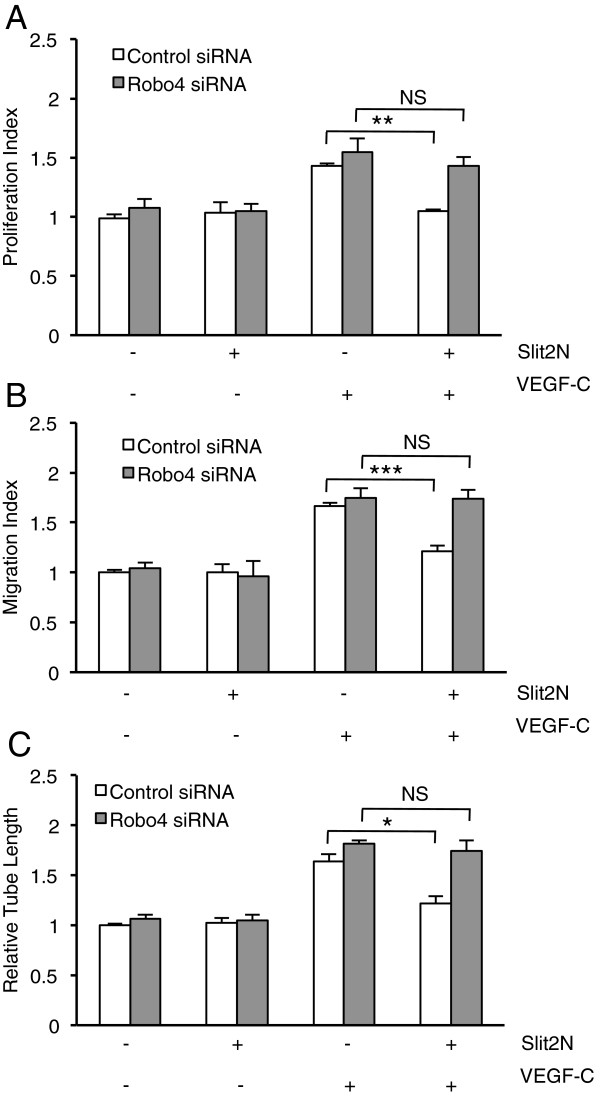
Results: We found that pretreatment with Slit2N, an active fragment of Slit2, inhibited VEGF-C-mediated lung-derived lymphatic endothelial cell (L-LEC) proliferation, migration, and in vitro tube formation. Slit2N induced the internalization of VEGFR-3, which blocked its activation, and inhibited the activation of the PI3K/Akt pathway by VEGF-C in L-LECs. Moreover, we found that inhibition of VEGF-C-induced effects by Slit2N was Robo4-dependent.
Conclusion: These results indicate that Slit2N/Robo4 modulates several key cellular functions, which contribute to lymphangiogenesis, and identify this ligand-receptor pair as a potential therapeutic target to inhibit lymphatic metastasis of VEGF-C-overexpressing cancers and manage lymphatic dysfunctions characterized by VEGF-C/VEGFR-3 activation.
Figures






Similar articles
-
Roundabout4 suppresses glioma-induced endothelial cell proliferation, migration and tube formation in vitro by inhibiting VEGR2-mediated PI3K/AKT and FAK signaling pathways.Cell Physiol Biochem. 2015;35(5):1689-705. doi: 10.1159/000373982. Epub 2015 Mar 18. Cell Physiol Biochem. 2015. PMID: 25833462
-
Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3.Breast Cancer Res. 2011 Jun 21;13(3):R66. doi: 10.1186/bcr2903. Breast Cancer Res. 2011. PMID: 21693010 Free PMC article.
-
Vascular endothelial growth factor receptor-3 directly interacts with phosphatidylinositol 3-kinase to regulate lymphangiogenesis.PLoS One. 2012;7(6):e39558. doi: 10.1371/journal.pone.0039558. Epub 2012 Jun 22. PLoS One. 2012. PMID: 22745786 Free PMC article.
-
Molecular control of lymphatic metastasis.Ann N Y Acad Sci. 2008;1131:225-34. doi: 10.1196/annals.1413.020. Ann N Y Acad Sci. 2008. PMID: 18519975 Review.
-
The role of the VEGF-C/VEGFR-3 axis in cancer progression.Br J Cancer. 2007 Feb 26;96(4):541-5. doi: 10.1038/sj.bjc.6603487. Epub 2006 Dec 12. Br J Cancer. 2007. PMID: 17164762 Free PMC article. Review.
Cited by
-
The lymphatic system and pancreatic cancer.Cancer Lett. 2016 Oct 10;381(1):217-36. doi: 10.1016/j.canlet.2015.11.048. Epub 2015 Dec 29. Cancer Lett. 2016. PMID: 26742462 Free PMC article. Review.
-
Emerging Role of the Slit/Roundabout (Robo) Signaling Pathway in Glioma Pathogenesis and Potential Therapeutic Options.Biomolecules. 2024 Sep 29;14(10):1231. doi: 10.3390/biom14101231. Biomolecules. 2024. PMID: 39456164 Free PMC article. Review.
-
Regulation of VEGFR3 signaling in lymphatic endothelial cells.Front Cell Dev Biol. 2025 Feb 13;13:1527971. doi: 10.3389/fcell.2025.1527971. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40046235 Free PMC article. Review.
-
HIV-associated preeclampsia: evaluation of lymphangiogenesis in placental bed samples.Histochem Cell Biol. 2025 May 22;163(1):55. doi: 10.1007/s00418-025-02359-4. Histochem Cell Biol. 2025. PMID: 40402284 Free PMC article.
-
Angiogenesis, Lymphangiogenesis, and the Immune Response in South African Preeclamptic Women Receiving HAART.Int J Mol Sci. 2019 Jul 30;20(15):3728. doi: 10.3390/ijms20153728. Int J Mol Sci. 2019. PMID: 31366152 Free PMC article. Review.
References
-
- Bahram F, Claesson-Welsh L. VEGF-mediated signal transduction in lymphatic endothelial cells. Pathophysiology: the official journal of the International Society for Pathophysiology/ISP. 2010;17:253–261. - PubMed
-
- Salameh A, Galvagni F, Bardelli M, Bussolino F, Oliviero S. Direct recruitment of CRK and GRB2 to VEGFR-3 induces proliferation, migration, and survival of endothelial cells through the activation of ERK, AKT, and JNK pathways. Blood. 2005;106:3423–3431. doi: 10.1182/blood-2005-04-1388. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
