

CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction
- PMID: 24708850
- PMCID: PMC4021573
- DOI: 10.1186/2049-2618-2-11
CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction
Abstract
Background: Culture-independent molecular surveys targeting conserved marker genes, most notably 16S rRNA, to assess microbial diversity remain semi-quantitative due to variations in the number of gene copies between species.
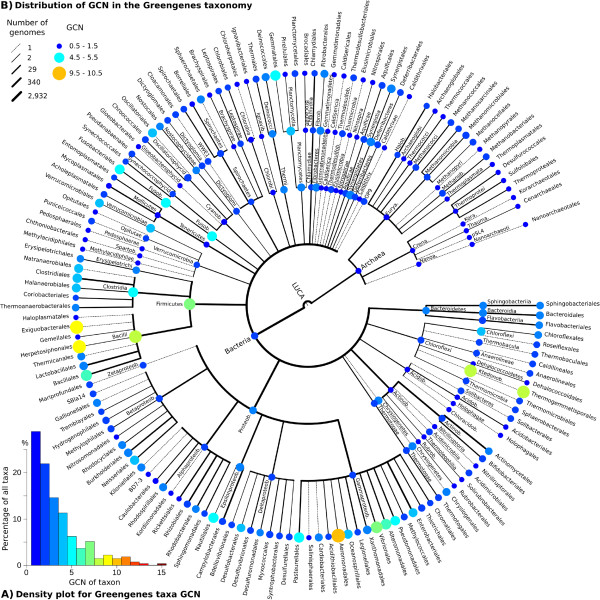
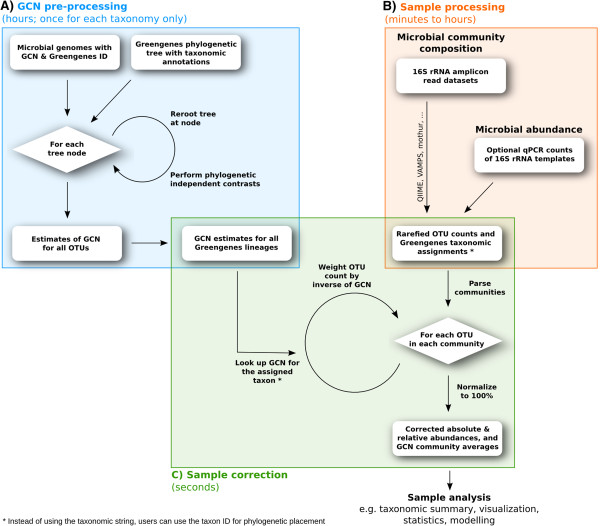
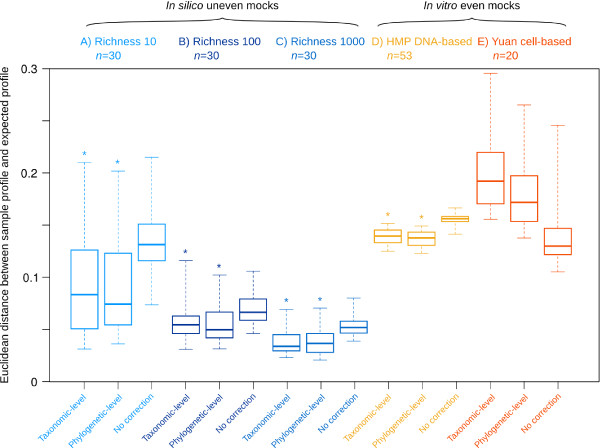
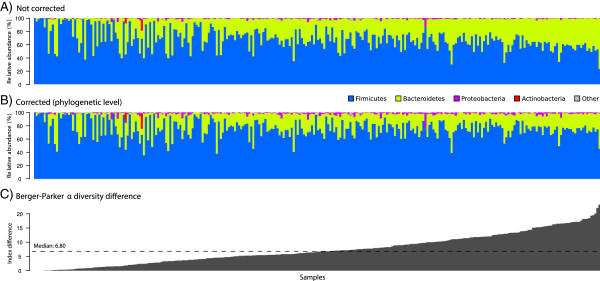
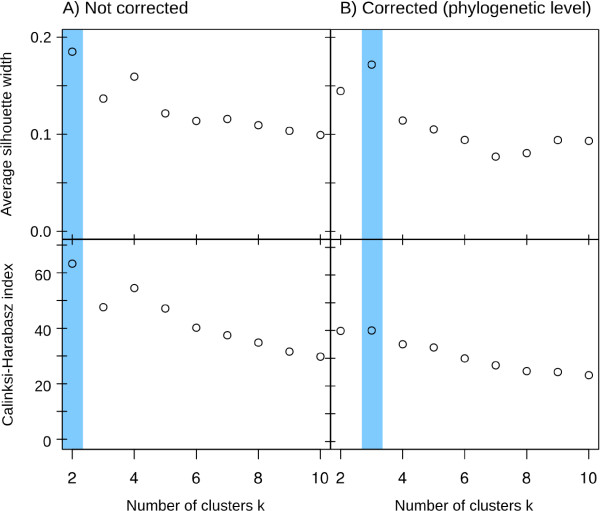
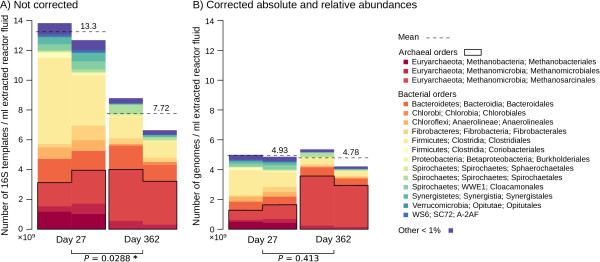
Results: Based on 2,900 sequenced reference genomes, we show that 16S rRNA gene copy number (GCN) is strongly linked to microbial phylogenetic taxonomy, potentially under-representing Archaea in amplicon microbial profiles. Using this relationship, we inferred the GCN of all bacterial and archaeal lineages in the Greengenes database within a phylogenetic framework. We created CopyRighter, new software which uses these estimates to correct 16S rRNA amplicon microbial profiles and associated quantitative (q)PCR total abundance. CopyRighter parses microbial profiles and, because GCN estimates are pre-computed for all taxa in the reference taxonomy, rapidly corrects GCN bias. Software validation with in silico and in vitro mock communities indicated that GCN correction results in more accurate estimates of microbial relative abundance and improves the agreement between metagenomic and amplicon profiles. Analyses of human-associated and anaerobic digester microbiomes illustrate that correction makes tangible changes to estimates of qPCR total abundance, α and β diversity, and can significantly change biological interpretation. For example, human gut microbiomes from twins were reclassified into three rather than two enterotypes after GCN correction.
Conclusions: The CopyRighter bioinformatic tools permits rapid correction of GCN in microbial surveys, resulting in improved estimates of microbial abundance, α and β diversity.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
