

Reassessing domain architecture evolution of metazoan proteins: major impact of errors caused by confusing paralogs and epaktologs
- PMID: 24710209
- PMCID: PMC3927612
- DOI: 10.3390/genes2030516
Reassessing domain architecture evolution of metazoan proteins: major impact of errors caused by confusing paralogs and epaktologs
Abstract
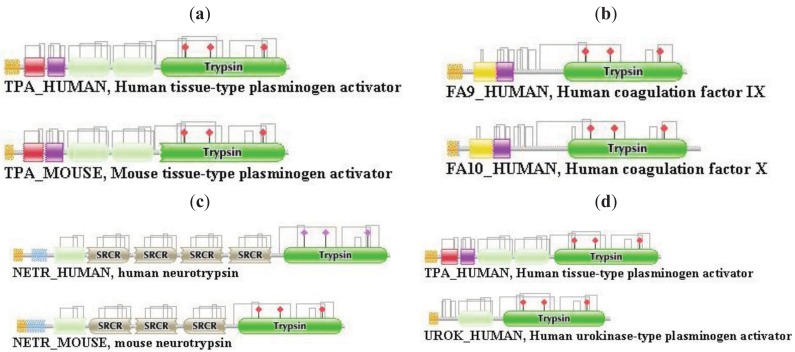
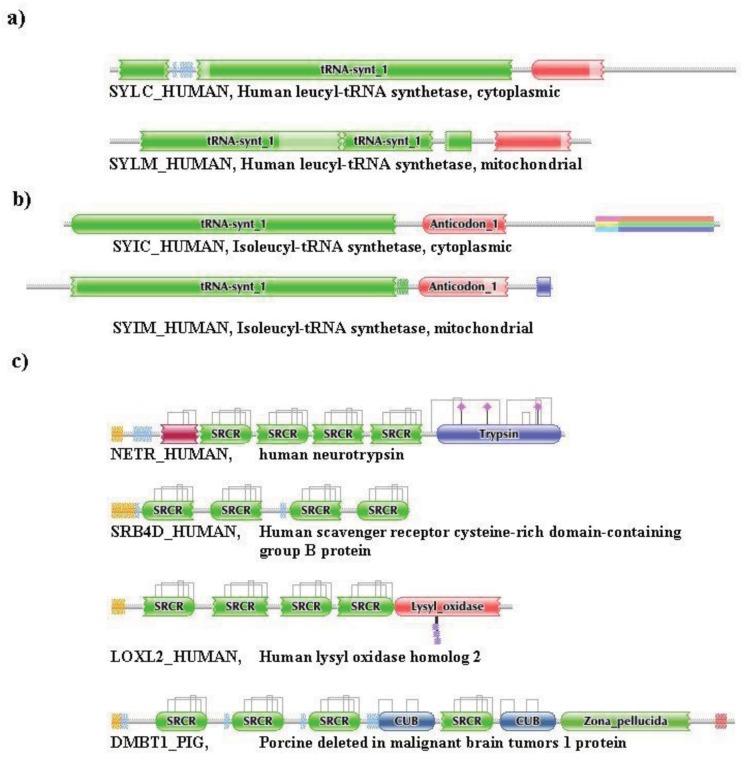
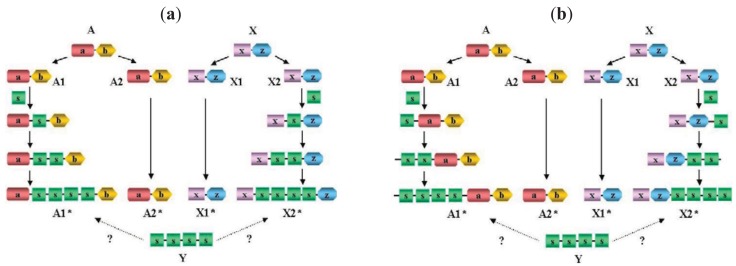
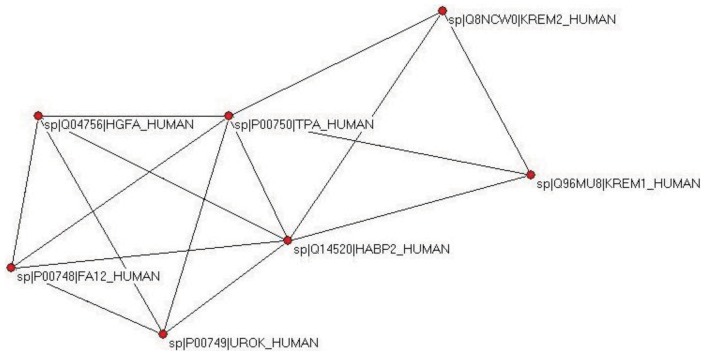
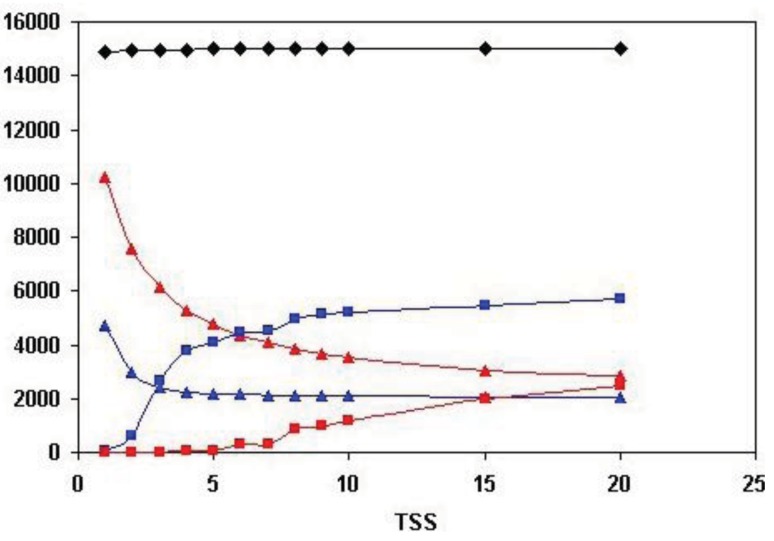
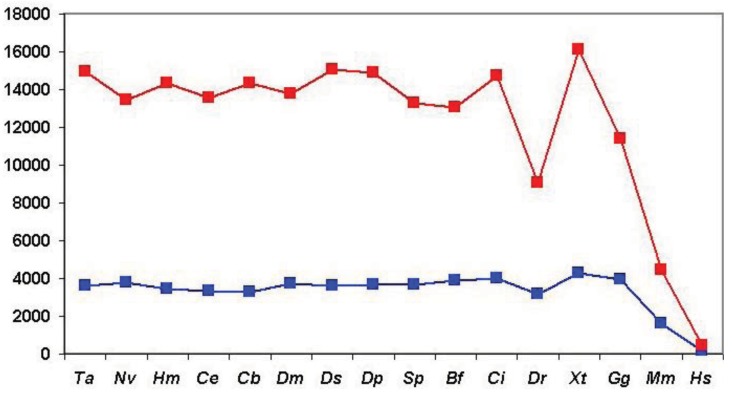
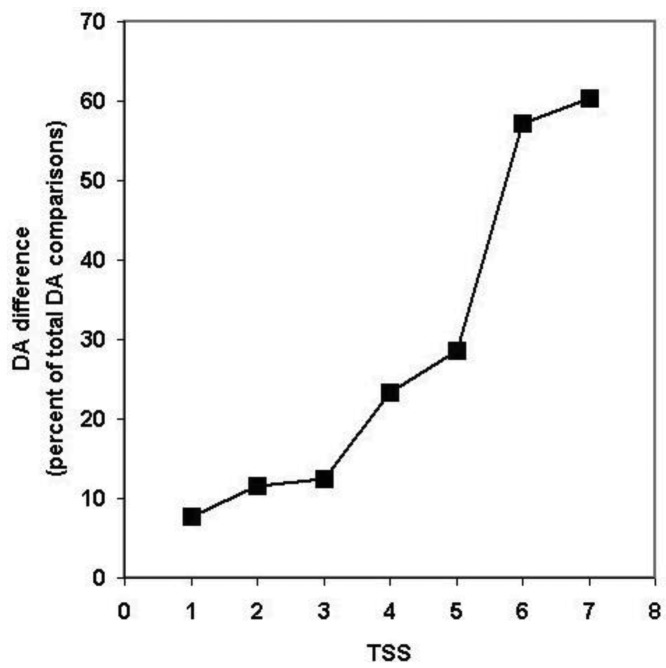
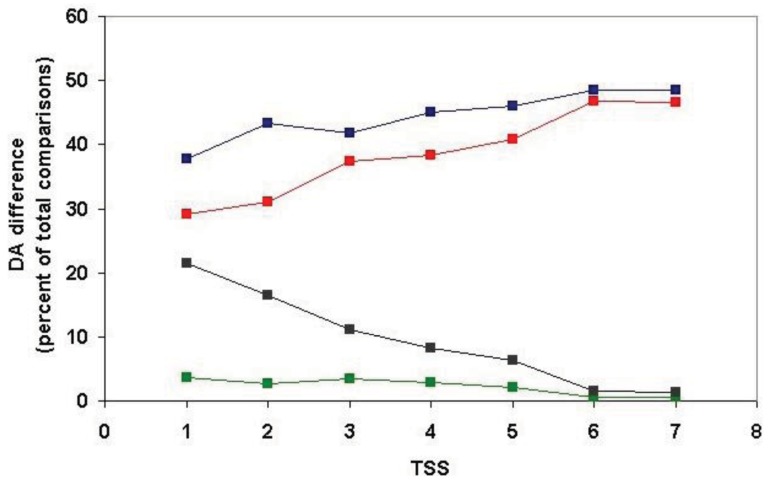
In the accompanying paper (Nagy, Szláma, Szarka, Trexler, Bányai, Patthy, Reassessing Domain Architecture Evolution of Metazoan Proteins: Major Impact of Gene Prediction Errors) we showed that in the case of UniProtKB/TrEMBL, RefSeq, EnsEMBL and NCBI's GNOMON predicted protein sequences of Metazoan species the contribution of erroneous (incomplete, abnormal, mispredicted) sequences to domain architecture (DA) differences of orthologous proteins might be greater than those of true gene rearrangements. Based on these findings, we suggest that earlier genome-scale studies based on comparison of predicted (frequently mispredicted) protein sequences may have led to some erroneous conclusions about the evolution of novel domain architectures of multidomain proteins. In this manuscript we examine the impact of confusing paralogous and epaktologous multidomain proteins (i.e., those that are related only through the independent acquisition of the same domain types) on conclusions drawn about DA evolution of multidomain proteins in Metazoa. To estimate the contribution of this type of error we have used as reference UniProtKB/Swiss-Prot sequences from protein families with well-characterized evolutionary histories. We have used two types of paralogy-group construction procedures and monitored the impact of various parameters on the separation of true paralogs from epaktologs on correctly annotated Swiss-Prot entries of multidomain proteins. Our studies have shown that, although public protein family databases are contaminated with epaktologs, analysis of the structure of sequence similarity networks of multidomain proteins provides an efficient means for the separation of epaktologs and paralogs. We have also demonstrated that contamination of protein families with epaktologs increases the apparent rate of DA change and introduces a bias in DA differences in as much as it increases the proportion of terminal over internal DA differences.We have shown that confusing paralogous and epaktologous multidomain proteins significantly increases the apparent rate of DA change in Metazoa and introduces a positional bias in favor of terminal over internal DA changes. Our findings caution that earlier studies based on analysis of datasets of protein families that were contaminated with epaktologs may have led to some erroneous conclusions about the evolution of novel domain architectures of multidomain proteins. A reassessment of the DA evolution of multidomain proteins is presented in an accompanying paper [1].
Figures
Similar articles
-
Reassessing domain architecture evolution of metazoan proteins: the contribution of different evolutionary mechanisms.Genes (Basel). 2011 Aug 5;2(3):578-98. doi: 10.3390/genes2030578. Genes (Basel). 2011. PMID: 24710211 Free PMC article.
-
Reassessing domain architecture evolution of metazoan proteins: major impact of gene prediction errors.Genes (Basel). 2011 Jul 13;2(3):449-501. doi: 10.3390/genes2030449. Genes (Basel). 2011. PMID: 24710207 Free PMC article.
-
Identification and correction of abnormal, incomplete and mispredicted proteins in public databases.BMC Bioinformatics. 2008 Aug 27;9:353. doi: 10.1186/1471-2105-9-353. BMC Bioinformatics. 2008. PMID: 18752676 Free PMC article.
-
Evolution of protein domain architectures.Methods Mol Biol. 2012;856:187-216. doi: 10.1007/978-1-61779-585-5_8. Methods Mol Biol. 2012. PMID: 22399460 Review.
-
Genome evolution and the evolution of exon-shuffling--a review.Gene. 1999 Sep 30;238(1):103-14. doi: 10.1016/s0378-1119(99)00228-0. Gene. 1999. PMID: 10570989 Review.
Cited by
-
Morphological Stasis and Proteome Innovation in Cephalochordates.Genes (Basel). 2018 Jul 16;9(7):353. doi: 10.3390/genes9070353. Genes (Basel). 2018. PMID: 30013013 Free PMC article.
-
The role of public goods in planetary evolution.Philos Trans A Math Phys Eng Sci. 2017 Dec 28;375(2109):20160359. doi: 10.1098/rsta.2016.0359. Philos Trans A Math Phys Eng Sci. 2017. PMID: 29133456 Free PMC article.
-
Putative extremely high rate of proteome innovation in lancelets might be explained by high rate of gene prediction errors.Sci Rep. 2016 Aug 1;6:30700. doi: 10.1038/srep30700. Sci Rep. 2016. PMID: 27476717 Free PMC article.
-
Probing the boundaries of orthology: the unanticipated rapid evolution of Drosophila centrosomin.Genetics. 2013 Aug;194(4):903-26. doi: 10.1534/genetics.113.152546. Epub 2013 Jun 7. Genetics. 2013. PMID: 23749319 Free PMC article.
-
Reassessing domain architecture evolution of metazoan proteins: the contribution of different evolutionary mechanisms.Genes (Basel). 2011 Aug 5;2(3):578-98. doi: 10.3390/genes2030578. Genes (Basel). 2011. PMID: 24710211 Free PMC article.
References
-
- Patthy L. Modular assembly of genes and the evolution of new functions. Genetica. 2003;118:217–231. - PubMed
-
- Tordai H., Nagy A., Farkas K., Banyai L., Patthy L. Modules, multidomain proteins and organismic complexity. FEBS J. 2005;272:5064–5078. - PubMed
-
- Fitch W.M. Homology: A personal view on some of the problems. Trends Genet. 2000;16:227–231. - PubMed
LinkOut - more resources
Full Text Sources
