

Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation
- PMID: 24711457
- PMCID: PMC4031511
- DOI: 10.1074/jbc.M114.564674
Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation
Abstract
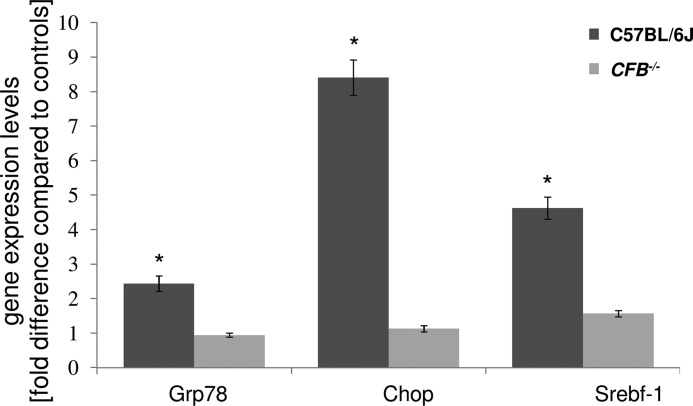
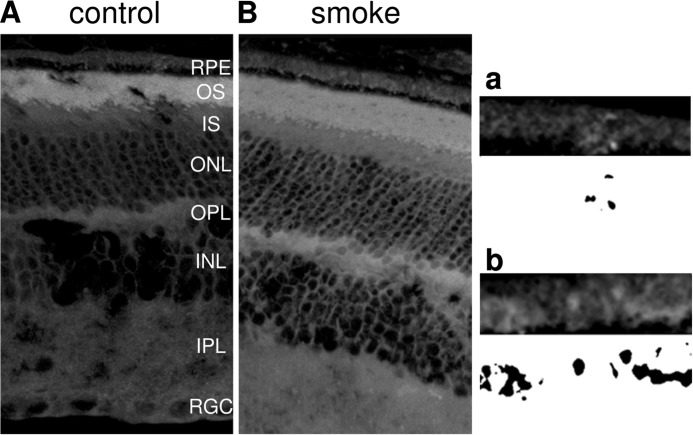
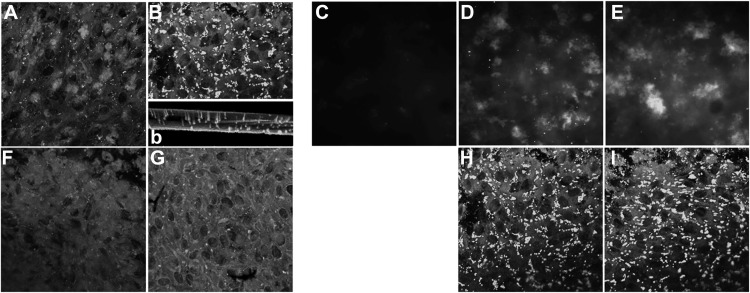
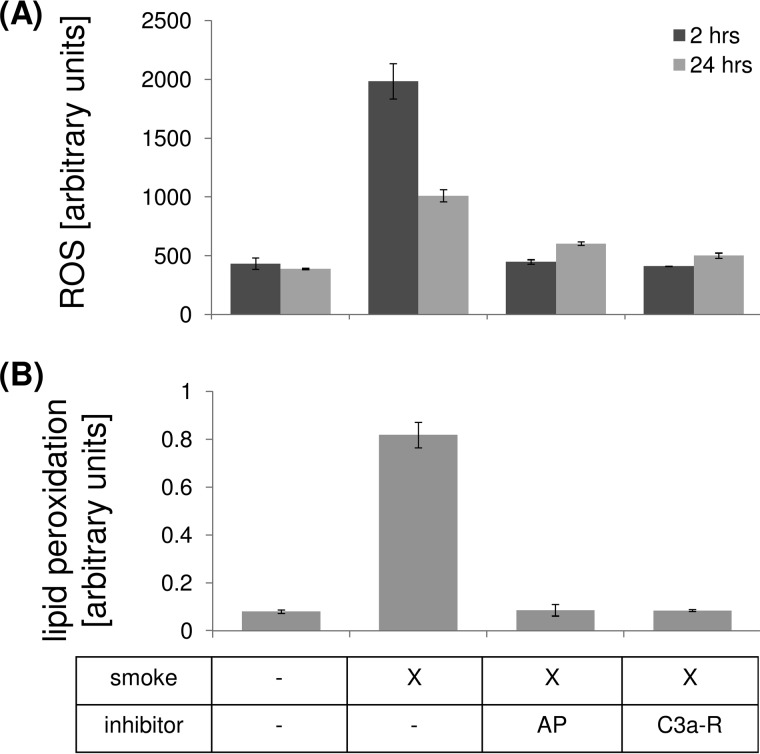
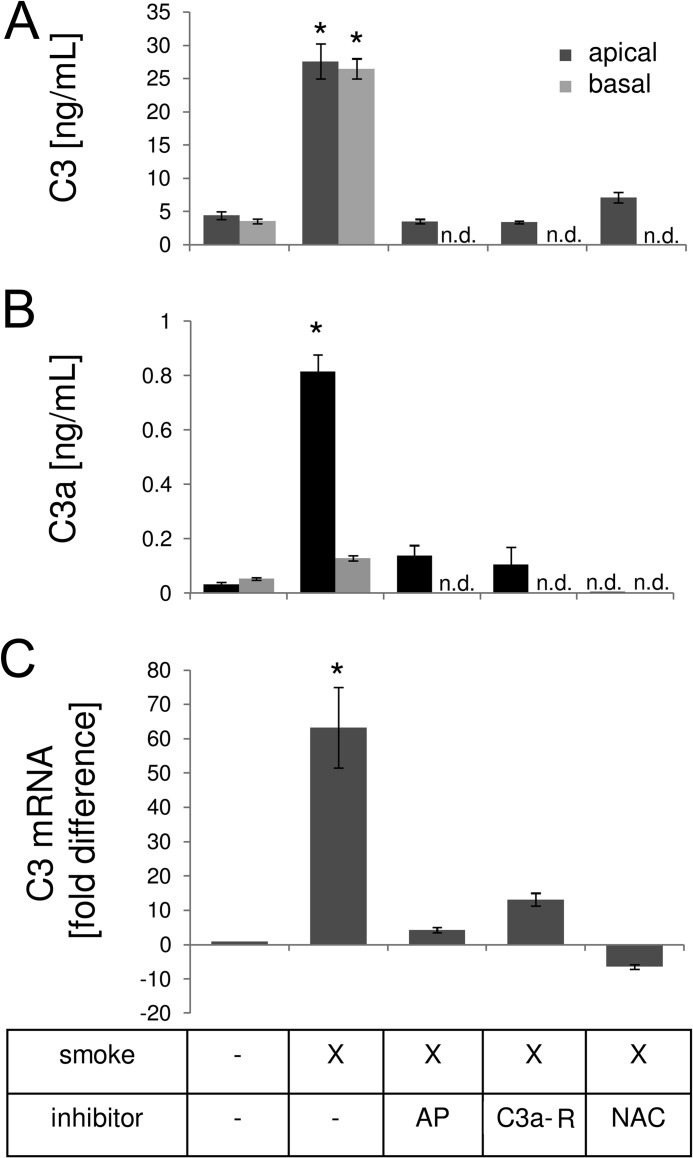
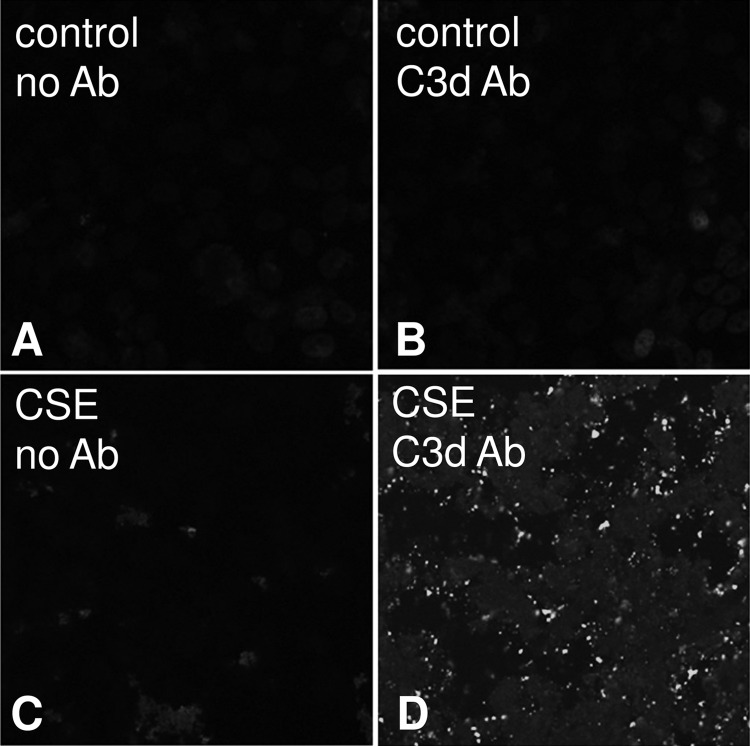
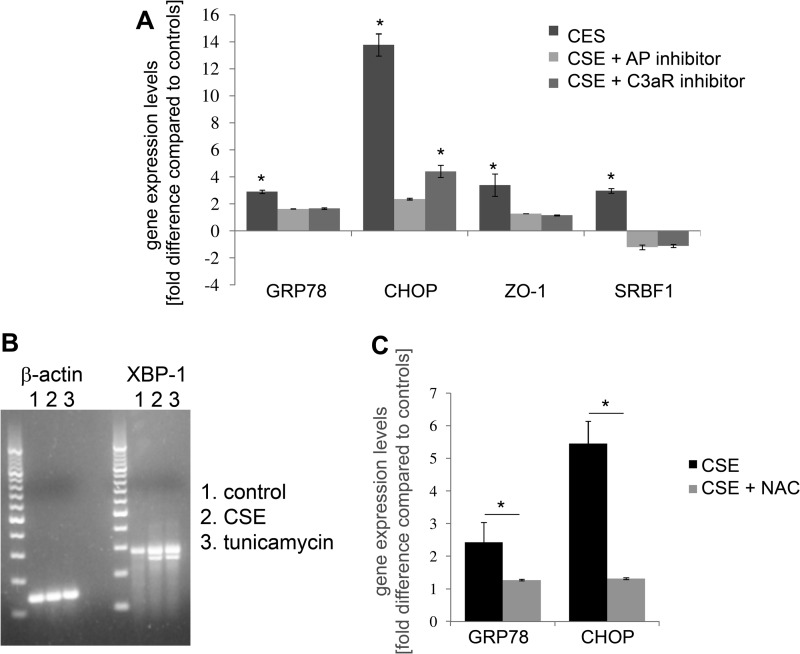
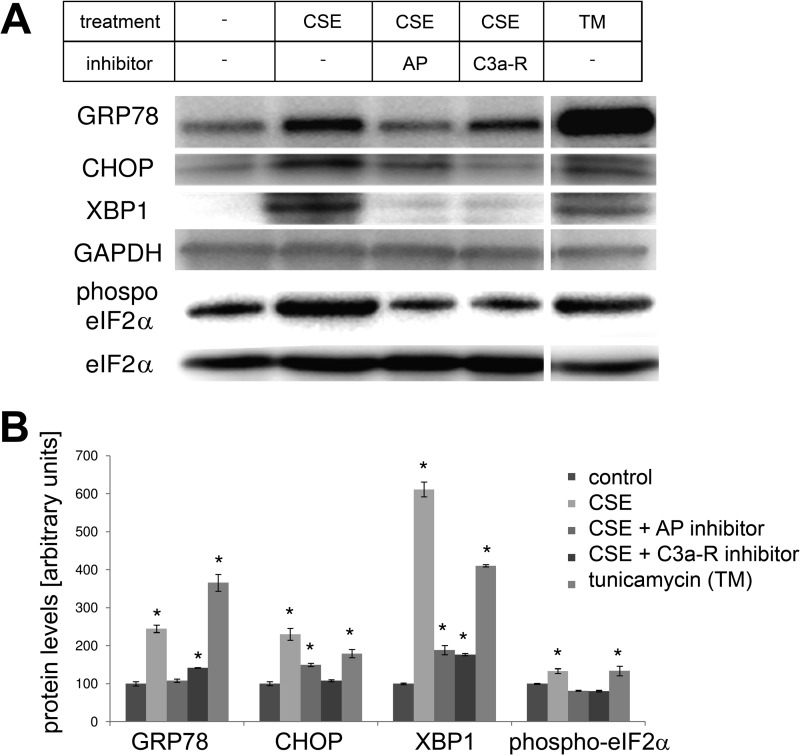
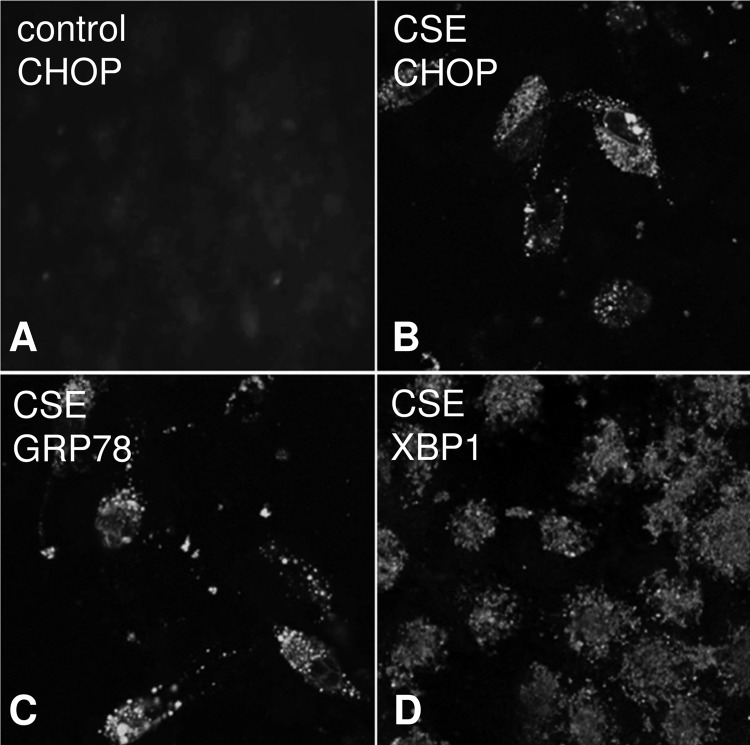
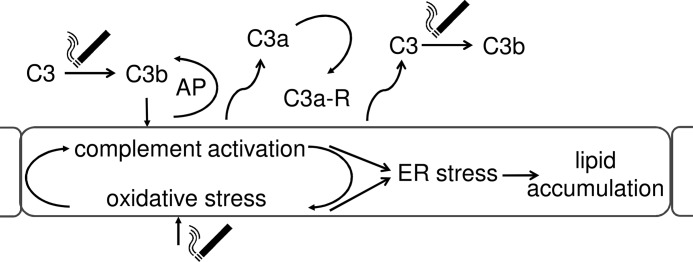
Age-related macular degeneration (AMD) is a complex disease caused by genetic and environmental factors, including genetic variants in complement components and smoking. Smoke exposure leads to oxidative stress, complement activation, endoplasmic reticulum (ER) stress, and lipid dysregulation, which have all been proposed to be associated with AMD pathogenesis. Here we examine the effects of smoke exposure on the retinal pigment epithelium (RPE). Mice were exposed to cigarette smoke or filtered air for 6 months. RPE cells grown as stable monolayers were exposed to 5% cigarette smoke extract (CSE). Effects of smoke were determined by biochemical, molecular, and histological measures. Effects of the alternative pathway (AP) of complement and complement C3a anaphylatoxin receptor signaling were analyzed using knock-out mice or specific inhibitors. ER stress markers were elevated after smoke exposure in RPE of intact mice, which was eliminated in AP-deficient mice. To examine this relationship further, RPE monolayers were exposed to CSE. Short term smoke exposure resulted in production and release of complement C3, the generation of C3a, oxidative stress, complement activation on the cell membrane, and ER stress. Long term exposure to CSE resulted in lipid accumulation, and secretion. All measures were reversed by blocking C3a complement receptor (C3aR), alternative complement pathway signaling, and antioxidant therapy. Taken together, our results provide clear evidence that smoke exposure results in oxidative stress and complement activation via the AP, resulting in ER stress-mediated lipid accumulation, and further suggesting that oxidative stress and complement act synergistically in the pathogenesis of AMD.
Keywords: Age-related Macular Degeneration; Complement System; ER Stress; Lipodystrophy; Mitochondria; Oxidative Stress; Smoking; Unfolded Protein Response.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Brown M. M., Brown G. C., Stein J. D., Roth Z., Campanella J., Beauchamp G. R. (2005) Age-related macular degeneration: economic burden and value-based medicine analysis. Can. J. Ophthalmol. 40, 277–287 - PubMed
-
- Freund K. B., Zweifel S. A., Engelbert M. (2010) Do we need a new classification for choroidal neovascularization in age-related macular degeneration? Retina 30, 1333–1349 - PubMed
-
- Hageman G. S., Luthert P. J., Victor Chong N. H., Johnson L. V., Anderson D. H., Mullins R. F. (2001) An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 20, 705–732 - PubMed
-
- Edwards A. O., Ritter R., 3rd, Abel K. J., Manning A., Panhuysen C., Farrer L. A. (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
