

Mass spectrometry based biomarker discovery, verification, and validation--quality assurance and control of protein biomarker assays
- PMID: 24713096
- PMCID: PMC5528535
- DOI: 10.1016/j.molonc.2014.03.006
Mass spectrometry based biomarker discovery, verification, and validation--quality assurance and control of protein biomarker assays
Abstract
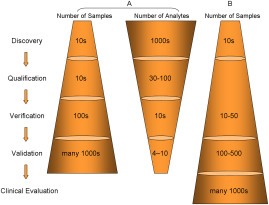
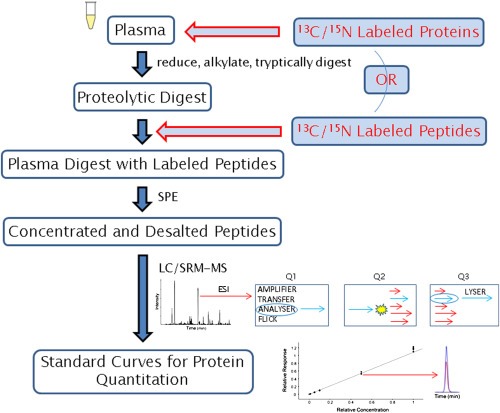
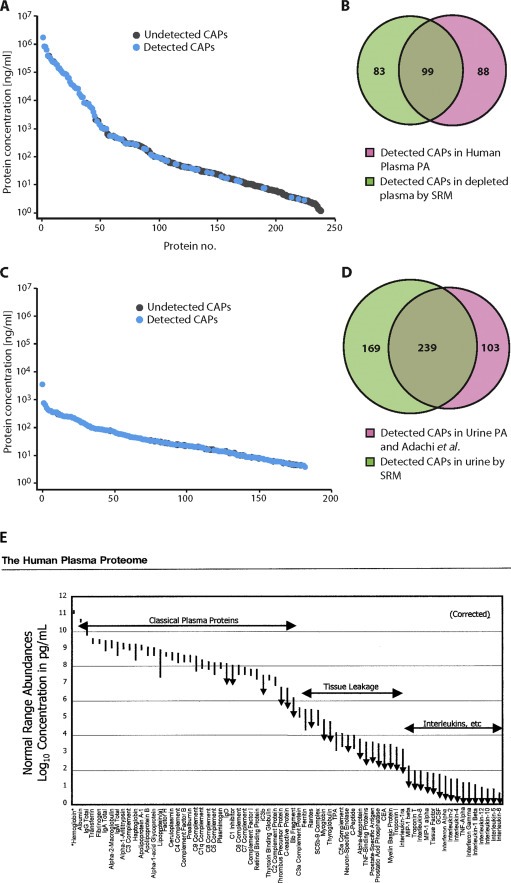
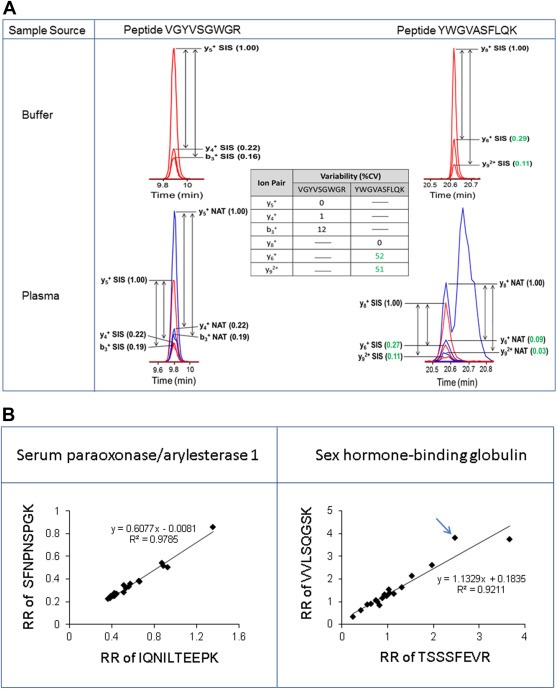
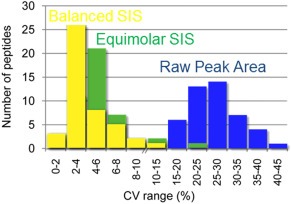
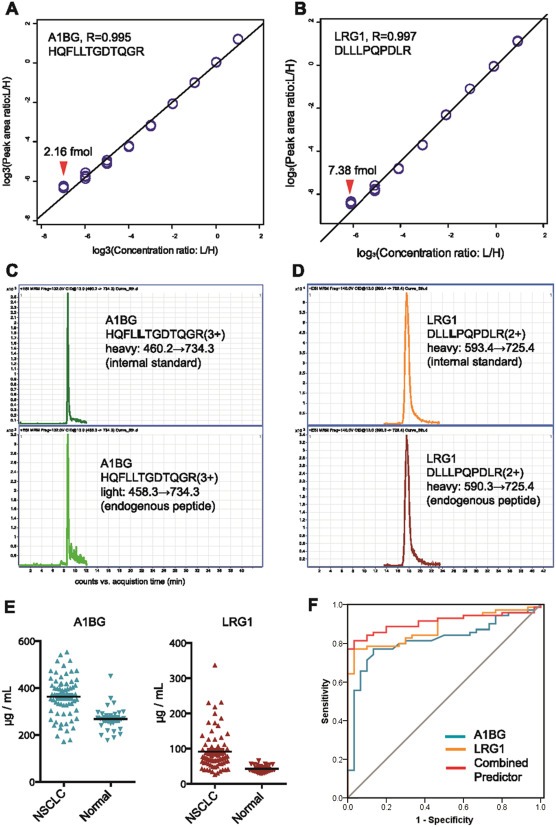
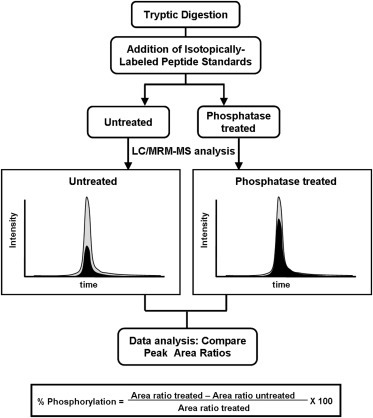
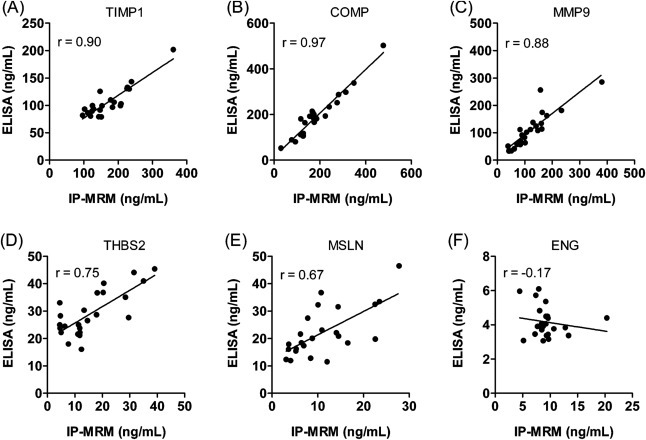
In its early years, mass spectrometry (MS)-based proteomics focused on the cataloging of proteins found in different species or different tissues. By 2005, proteomics was being used for protein quantitation, typically based on "proteotypic" peptides which act as surrogates for the parent proteins. Biomarker discovery is usually done by non-targeted "shotgun" proteomics, using relative quantitation methods to determine protein expression changes that correlate with disease (output given as "up-or-down regulation" or "fold-increases"). MS-based techniques can also perform "absolute" quantitation which is required for clinical applications (output given as protein concentrations). Here we describe the differences between these methods, factors that affect the precision and accuracy of the results, and some examples of recent studies using MS-based proteomics to verify cancer-related biomarkers.
Keywords: Biomarker discovery; Biomarkers; Cancer; Mass spectrometry; Multiple reaction monitoring; Plasma or serum; Selected reaction monitoring; Targeted proteomics; Validation; Verification.
Copyright © 2014 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
Figures
Similar articles
-
A large-scale and robust dynamic MRM study of colorectal cancer biomarkers.J Proteomics. 2018 Sep 15;187:80-92. doi: 10.1016/j.jprot.2018.06.013. Epub 2018 Jun 25. J Proteomics. 2018. PMID: 29953963
-
Delineating protein biomarkers for gastric cancers: A catalogue of mass spectrometry-based markers and assessment of their suitability for targeted proteomics applications.J Proteomics. 2024 Aug 30;306:105262. doi: 10.1016/j.jprot.2024.105262. Epub 2024 Jul 22. J Proteomics. 2024. PMID: 39047941
-
Absolute Quantification of Middle- to High-Abundant Plasma Proteins via Targeted Proteomics.Methods Mol Biol. 2017;1619:417-430. doi: 10.1007/978-1-4939-7057-5_29. Methods Mol Biol. 2017. PMID: 28674901
-
Targeted mass spectrometry approaches for protein biomarker verification.J Proteomics. 2011 Nov 18;74(12):2650-9. doi: 10.1016/j.jprot.2011.04.011. Epub 2011 Apr 21. J Proteomics. 2011. PMID: 21540133 Review.
-
SRM/MRM targeted proteomics as a tool for biomarker validation and absolute quantification in human urine.Expert Rev Mol Diagn. 2015;15(11):1441-54. doi: 10.1586/14737159.2015.1093937. Epub 2015 Oct 15. Expert Rev Mol Diagn. 2015. PMID: 26472065 Review.
Cited by
-
Mass spectroscopy-based proteomics and metabolomics analysis of triple-positive breast cancer cells treated with tamoxifen and/or trastuzumab.Cancer Chemother Pharmacol. 2022 Dec;90(6):467-488. doi: 10.1007/s00280-022-04478-4. Epub 2022 Oct 20. Cancer Chemother Pharmacol. 2022. PMID: 36264351
-
Oxyntomodulin Identified as a Marker of Type 2 Diabetes and Gastric Bypass Surgery by Mass-spectrometry Based Profiling of Human Plasma.EBioMedicine. 2016 May;7:112-20. doi: 10.1016/j.ebiom.2016.03.034. Epub 2016 Mar 31. EBioMedicine. 2016. PMID: 27322465 Free PMC article.
-
Clinical Validation of a Serum Protein Panel (FLNA, FLNB and KRT19) for Diagnosis of Prostate Cancer.J Mol Biomark Diagn. 2017;8(2):323. doi: 10.4172/2155-9929.1000323. Epub 2017 Feb 8. J Mol Biomark Diagn. 2017. PMID: 29682400 Free PMC article.
-
Assuring Consistent Performance of an Insulin-Like Growth Factor 1 MALDImmunoassay by Monitoring Measurement Quality Indicators.Anal Chem. 2017 Jun 6;89(11):6188-6195. doi: 10.1021/acs.analchem.7b01125. Epub 2017 May 10. Anal Chem. 2017. PMID: 28467045 Free PMC article.
-
Modeling and integration of N-glycan biomarkers in a comprehensive biomarker data model.Glycobiology. 2022 Sep 19;32(10):855-870. doi: 10.1093/glycob/cwac046. Glycobiology. 2022. PMID: 35925813 Free PMC article.
References
-
- Abbatiello, S.E. , Mani, D.R. , Schilling, B. , Maclean, B. , Zimmerman, L.J. , Feng, X. , Cusack, M.P. , Sedransk, N. , Hall, S.C. , Addona, T. , Allen, S. , Dodder, N.G. , Ghosh, M. , Held, J.M. , V., H. , Inerowicz, H.D. , Jackson, A. , Keshishian, H. , Kim, J.W. , Lyssand, J.S. , Riley, C.P. , Rudnick, P. , Sadowski, P. , Shaddox, K. , Smith, D. , Tomazela, D. , Wahlander, A. , Waldemarson, S. , Whitwell, C.A. , You, J. , Zhang, S. , Kinsinger, C.R. , Mesri, M. , Rodriguez, H. , Borchers, C.H. , Buck, C. , Fisher, S.J. , Gibson, B.W. , Liebler, D. , Maccoss, M. , Neubert, T.A. , Paulovich, A. , Regnier, F. , Skates, S.J. , Tempst, P. , Wang, M. , Carr, S.A. , 2013. Design, implementation, and multi-site evaluation of a system suitability protocol for the quantitative assessment of instrument performance in LC-MRM-MS. Mol. Cell Proteomics. 12, 2623–2639. - PMC - PubMed
-
- Addona, T.A. , Abbatiello, S.E. , Schilling, B. , Skates, S.J. , Mani, D.R. , Bunk, D.M. , Spiegelman, C.H. , Zimmerman, L.J. , Ham, A.-J.L. , Keshishian, H. , Hall, S.C. , Allen, S. , Blackman, R.K. , Borchers, C.H. , Buck, C. , Cardasis, H.L. , Cusack, M.P. , Dodder, N.G. , Gibson, B.W. , Held, J.M. , Hiltke, T. , Jackson, A. , Johansen, E.B. , Kinsinger, C.R. , Li, J. , Mesri, M. , Neubert, T.A. , Niles, R.K. , Pulsipher, T.C. , Ransohoff, D. , Rodriguez, H. , Rudnick, P.A. , Smith, D. , Tabb, D.L. , Tegeler, T.J. , Variyath, A.M. , Vega-Montoto, L.J. , Wahlander, A. , Waldemarson, S. , Wang, M. , Whiteaker, J.R. , Zhao, L. , Anderson, N.L. , Fisher, S.J. , Liebler, D.C. , Paulovich, A.G. , Regnier, F.E. , Tempst, P. , Carr, S.A. , 2009. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol.. 27, 633–641. - PMC - PubMed
-
- Addona, T.A. , Shi, X. , Keshishian, H. , Mani, D.R. , Burgess, M. , Gillette, M.A. , Clauser, K.R. , Shen, D. , Lewis, G.D. , Farrell, L.A. , Fifer, M.A. , Sabatine, M.S. , Gerszten, R.E. , Carr, S.A. , 2011. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol.. 29, 635–643. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
