

Identification of Nine New RAI1-Truncating Mutations in Smith-Magenis Syndrome Patients without 17p11.2 Deletions
- PMID: 24715852
- PMCID: PMC3977224
- DOI: 10.1159/000357359
Identification of Nine New RAI1-Truncating Mutations in Smith-Magenis Syndrome Patients without 17p11.2 Deletions
Abstract
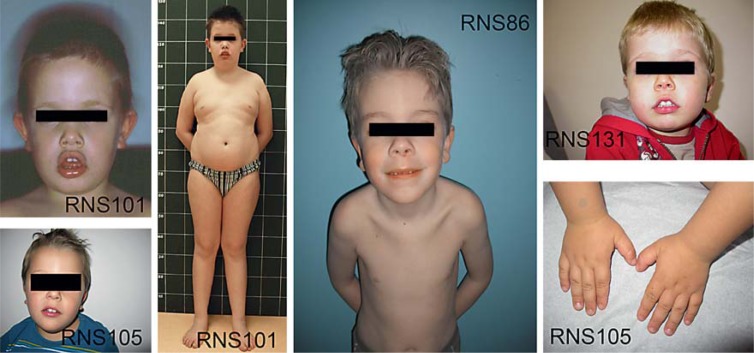
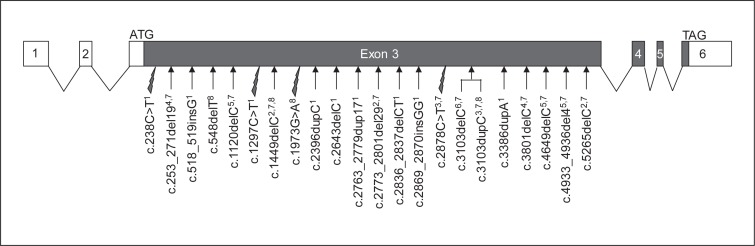
Smith-Magenis syndrome (SMS) is an intellectual disability syndrome with sleep disturbance, self-injurious behaviors and dysmorphic features. It is estimated to occur in 1/25,000 births, and in 90% of cases it is associated with interstitial deletions of chromosome 17p11.2. RAI1 (retinoic acid induced 1; OMIM 607642) mutations are the second most frequent molecular etiology, with this gene being located in the SMS locus at 17p11.2. Here, we report 9 new RAI1-truncating mutations in nonrelated individuals referred for molecular analysis due to a possible SMS diagnosis. None of these patients carried a 17p11.2 deletion. The 9 mutations include 2 nonsense mutations and 7 heterozygous frameshift mutations leading to protein truncation. All mutations map in exon 3 of RAI1 which codes for more than 98% of the protein. RAI1 regulates gene transcription, and its targets are themselves involved in transcriptional regulation, cell growth and cell cycle regulation, bone and skeletal development, lipid and glucide metabolisms, neurological development, behavioral functions, and circadian activity. We report the clinical features of the patients carrying these deleterious mutations in comparison with those of patients carrying 17p11.2 deletions.
Keywords: 17p11.2; Mutation; RAI1; Smith-Magenis syndrome.
Figures
Similar articles
-
RAI1 gene mutations: mechanisms of Smith-Magenis syndrome.Appl Clin Genet. 2017 Nov 3;10:85-94. doi: 10.2147/TACG.S128455. eCollection 2017. Appl Clin Genet. 2017. PMID: 29138588 Free PMC article. Review.
-
RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions.J Med Genet. 2005 Nov;42(11):820-8. doi: 10.1136/jmg.2005.031211. Epub 2005 Mar 23. J Med Genet. 2005. PMID: 15788730 Free PMC article.
-
Smith-Magenis Syndrome.2001 Oct 22 [updated 2025 May 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2001 Oct 22 [updated 2025 May 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301487 Free Books & Documents. Review.
-
Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum.Genet Med. 2006 Jul;8(7):417-27. doi: 10.1097/01.gim.0000228215.32110.89. Genet Med. 2006. PMID: 16845274
-
Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature.BMC Med Genet. 2010 Oct 8;11:142. doi: 10.1186/1471-2350-11-142. BMC Med Genet. 2010. PMID: 20932317 Free PMC article. Review.
Cited by
-
Whole exome sequencing identifies RAI1 mutation in a morbidly obese child diagnosed with ROHHAD syndrome.J Clin Endocrinol Metab. 2015 May;100(5):1723-30. doi: 10.1210/jc.2014-4215. Epub 2015 Mar 17. J Clin Endocrinol Metab. 2015. PMID: 25781356 Free PMC article.
-
Two Monogenetic Disorders, Activated PI3-Kinase-δ Syndrome 2 and Smith-Magenis Syndrome, in One Patient: Case Report and a Literature Review of Neurodevelopmental Impact in Primary Immunodeficiencies Associated With Disturbed PI3K Signaling.Front Pediatr. 2021 Jun 24;9:688022. doi: 10.3389/fped.2021.688022. eCollection 2021. Front Pediatr. 2021. PMID: 34249818 Free PMC article.
-
Novel RAI1:c.2736delC Variant in Smith-Magenis Syndrome: Identification by Whole Genome Sequencing and Joint Analysis.J Pers Med. 2024 Aug 25;14(9):901. doi: 10.3390/jpm14090901. J Pers Med. 2024. PMID: 39338155 Free PMC article.
-
RAI1 gene mutations: mechanisms of Smith-Magenis syndrome.Appl Clin Genet. 2017 Nov 3;10:85-94. doi: 10.2147/TACG.S128455. eCollection 2017. Appl Clin Genet. 2017. PMID: 29138588 Free PMC article. Review.
-
A case of Smith-Magenis syndrome with skin manifestations caused by a novel locus mutation in the RAI1 gene.J Int Med Res. 2023 Sep;51(9):3000605231190553. doi: 10.1177/03000605231190553. J Int Med Res. 2023. PMID: 37756600 Free PMC article.
References
-
- Antonarakis SE, Krawczak M, Cooper DN. Disease-causing mutations in the human genome. Eur J Pediatr 159 Suppl. 2000;3:S173–S178. - PubMed
-
- Bi W, Saifi GM, Shaw CJ, Walz K, Fonseca P, et al. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum Genet. 2004;115:515–524. - PubMed
-
- Bi W, Ohyama T, Nakamura H, Yan J, Visvanathan J, et al. Inactivation of Rai1 in mice recapitulates phenotypes observed in chromosome engineered mouse models for Smith-Magenis syndrome. Hum Mol Genet. 2005;14:983–995. - PubMed
-
- Bi W, Saifi GM, Girirajan S, Shi X, Szomju B, et al. RAI1 point mutations, CAG repeat variation, and SNP analysis in non-deletion Smith-Magenis syndrome. Am J Med Genet A. 2006;140:2454–2463. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
