

Mysteries of α1-antitrypsin deficiency: emerging therapeutic strategies for a challenging disease
- PMID: 24719116
- PMCID: PMC3974452
- DOI: 10.1242/dmm.014092
Mysteries of α1-antitrypsin deficiency: emerging therapeutic strategies for a challenging disease
Abstract
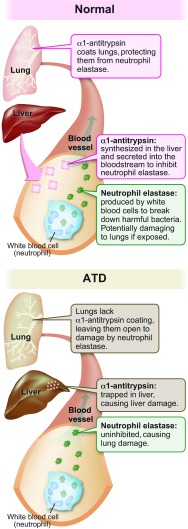
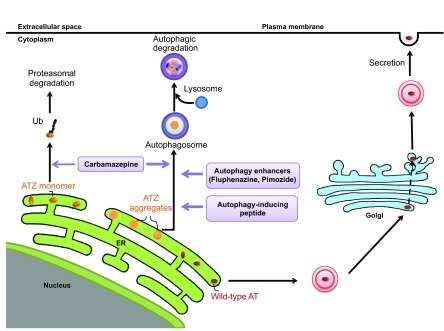
The classical form of α1-antitrypsin deficiency (ATD) is an autosomal co-dominant disorder that affects ~1 in 3000 live births and is an important genetic cause of lung and liver disease. The protein affected, α1-antitrypsin (AT), is predominantly derived from the liver and has the function of inhibiting neutrophil elastase and several other destructive neutrophil proteinases. The genetic defect is a point mutation that leads to misfolding of the mutant protein, which is referred to as α1-antitrypsin Z (ATZ). Because of its misfolding, ATZ is unable to efficiently traverse the secretory pathway. Accumulation of ATZ in the endoplasmic reticulum of liver cells has a gain-of-function proteotoxic effect on the liver, resulting in fibrosis, cirrhosis and/or hepatocellular carcinoma in some individuals. Moreover, because of reduced secretion, there is a lack of anti-proteinase activity in the lung, which allows neutrophil proteases to destroy the connective tissue matrix and cause chronic obstructive pulmonary disease (COPD) by loss of function. Wide variation in the incidence and severity of liver and lung disease among individuals with ATD has made this disease one of the most challenging of the rare genetic disorders to diagnose and treat. Other than cigarette smoking, which worsens COPD in ATD, genetic and environmental modifiers that determine this phenotypic variability are unknown. A limited number of therapeutic strategies are currently available, and liver transplantation is the only treatment for severe liver disease. Although replacement therapy with purified AT corrects the loss of anti-proteinase function, COPD progresses in a substantial number of individuals with ATD and some undergo lung transplantation. Nevertheless, advances in understanding the variability in clinical phenotype and in developing novel therapeutic concepts is beginning to address the major clinical challenges of this mysterious disorder.
Keywords: Autophagy; Liver disease; α1-antitrypsin deficiency.
Figures
References
-
- Alam S., Wang J., Janciauskiene S, Mahadeva R. (2012). Preventing and reversing the cellular consequences of Z alpha-1 antitrypsin accumulation by targeting s4A. J. Hepatol. 57, 116–124 - PubMed
-
- Alpha1-Antitrypsin Deficiency Registry Study Group (1994). A registry of patients with severe deficiency of alpha1-antitrypsin: Design and methods. Chest 106, 1223–1232 - PubMed
-
- Boglev Y., Badrock A. P., Trotter A. J., Du Q., Richardson E. J., Parslow A. C., Markmiller S. J., Hall N. E., de Jong-Curtain T. A., Ng A. Y., et al. (2013). Autophagy induction is a Tor- and Tp53-independent cell survival response in a zebrafish model of disrupted ribosome biogenesis. PLoS Genet. 9, e1003279. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
