

Inhibition of the cardiac Na⁺ channel Nav1.5 by carbon monoxide
- PMID: 24719320
- PMCID: PMC4047409
- DOI: 10.1074/jbc.M114.569996
Inhibition of the cardiac Na⁺ channel Nav1.5 by carbon monoxide
Abstract
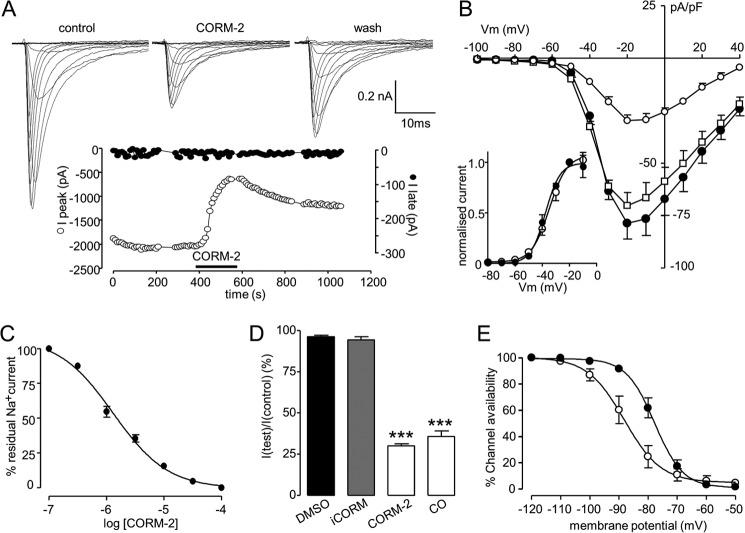
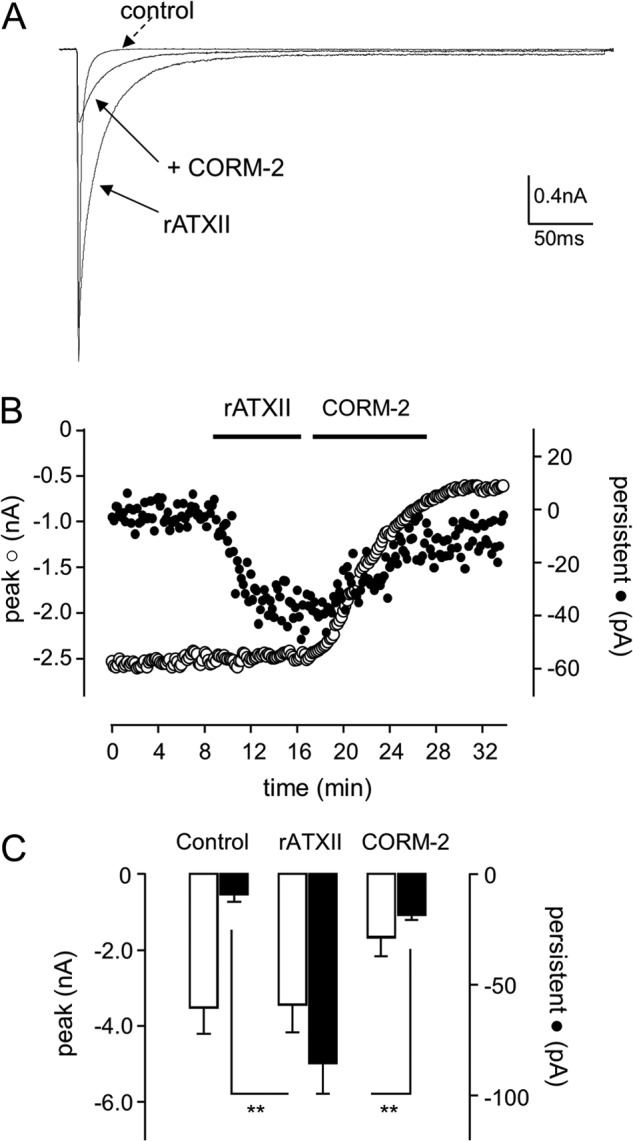
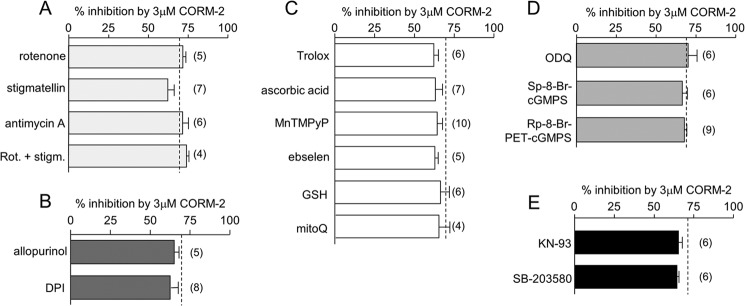
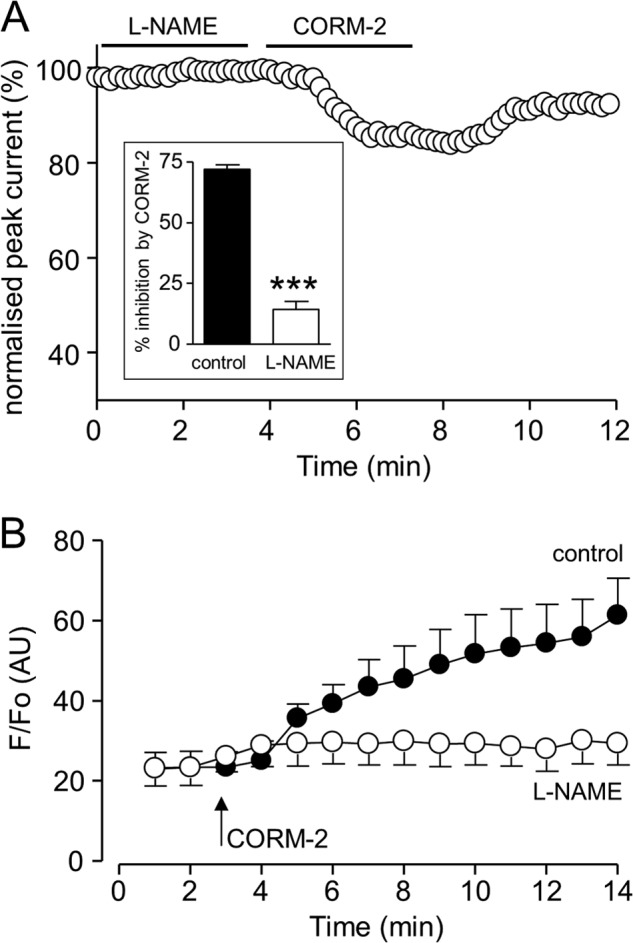
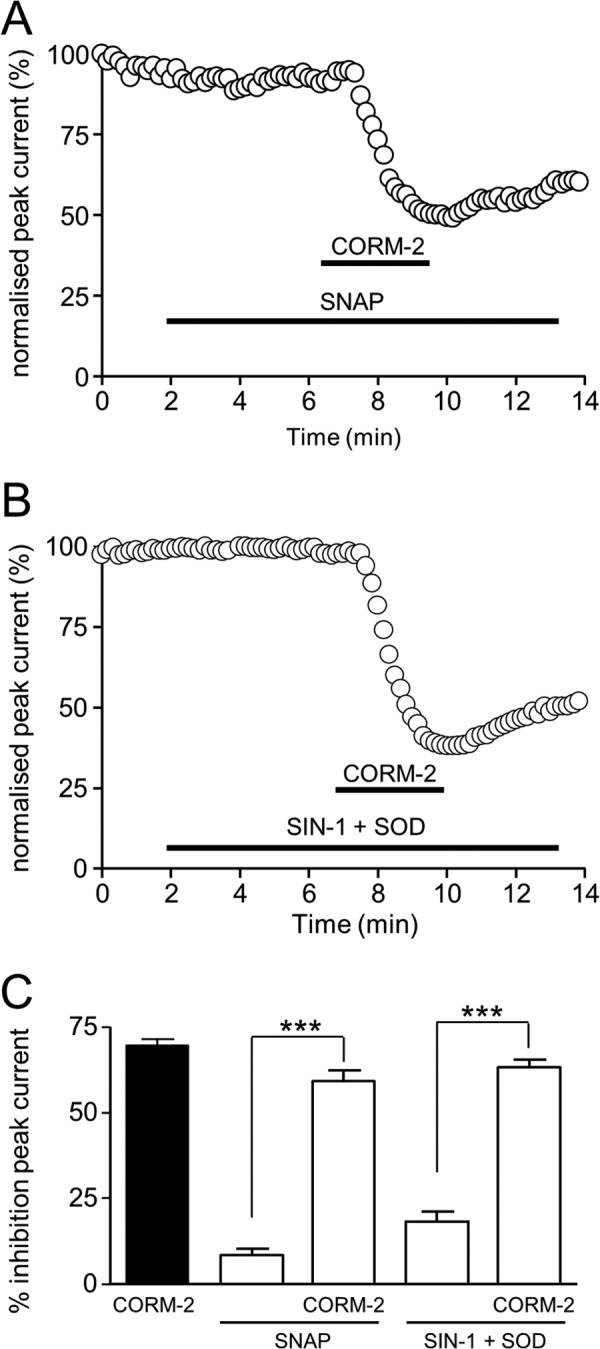
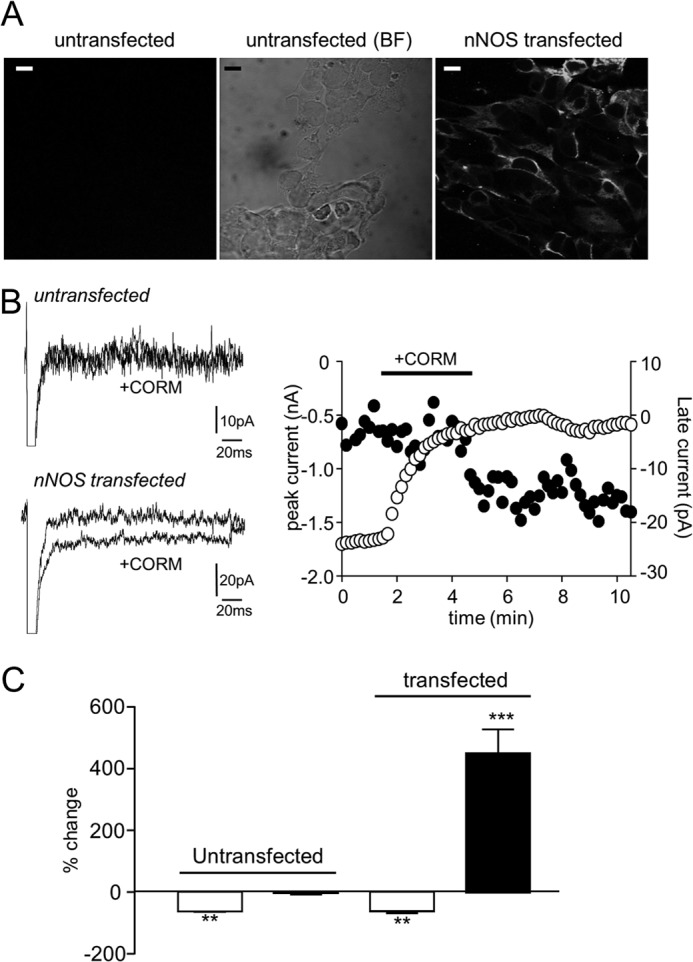
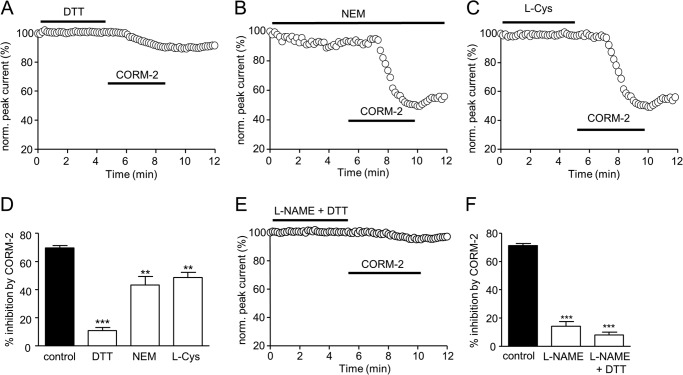
Sublethal carbon monoxide (CO) exposure is frequently associated with myocardial arrhythmias, and our recent studies have demonstrated that these may be attributable to modulation of cardiac Na(+) channels, causing an increase in the late current and an inhibition of the peak current. Using a recombinant expression system, we demonstrate that CO inhibits peak human Nav1.5 current amplitude without activation of the late Na(+) current observed in native tissue. Inhibition was associated with a hyperpolarizing shift in the steady-state inactivation properties of the channels and was unaffected by modification of channel gating induced by anemone toxin (rATX-II). Systematic pharmacological assessment indicated that no recognized CO-sensitive intracellular signaling pathways appeared to mediate CO inhibition of Nav1.5. Inhibition was, however, markedly suppressed by inhibition of NO formation, but NO donors did not mimic or occlude channel inhibition by CO, indicating that NO alone did not account for the actions of CO. Exposure of cells to DTT immediately before CO exposure also dramatically reduced the magnitude of current inhibition. Similarly, l-cysteine and N-ethylmaleimide significantly attenuated the inhibition caused by CO. In the presence of DTT and the NO inhibitor N(ω)-nitro-L-arginine methyl ester hydrochloride, the ability of CO to inhibit Nav1.5 was almost fully prevented. Our data indicate that inhibition of peak Na(+) current (which can lead to Brugada syndrome-like arrhythmias) occurs via a mechanism distinct from induction of the late current, requires NO formation, and is dependent on channel redox state.
Keywords: Arrhythmia; Carbon Monoxide; Heart; Nitric Oxide; Patch Clamp Electrophysiology; Sodium Channels.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Carbon monoxide induces cardiac arrhythmia via induction of the late Na+ current.Am J Respir Crit Care Med. 2012 Oct 1;186(7):648-56. doi: 10.1164/rccm.201204-0688OC. Epub 2012 Jul 19. Am J Respir Crit Care Med. 2012. PMID: 22822026 Free PMC article.
-
Mutant voltage-gated Na+ channels can exert a dominant negative effect through coupled gating.Am J Physiol Heart Circ Physiol. 2018 Nov 1;315(5):H1250-H1257. doi: 10.1152/ajpheart.00721.2017. Epub 2018 Aug 17. Am J Physiol Heart Circ Physiol. 2018. PMID: 30118344 Free PMC article.
-
The distinct effects of lipid emulsions used for "lipid resuscitation" on gating and bupivacaine-induced inhibition of the cardiac sodium channel Nav1.5.Anesth Analg. 2013 Nov;117(5):1101-8. doi: 10.1213/ANE.0b013e3182a1af78. Anesth Analg. 2013. PMID: 24029851
-
Hydrogen sulfide is a partially redox-independent activator of the human jejunum Na+ channel, Nav1.5.Am J Physiol Gastrointest Liver Physiol. 2011 Jun;300(6):G1105-14. doi: 10.1152/ajpgi.00556.2010. Epub 2011 Mar 10. Am J Physiol Gastrointest Liver Physiol. 2011. PMID: 21393430 Free PMC article.
-
Ca2+-dependent modulation of voltage-gated myocyte sodium channels.Biochem Soc Trans. 2021 Nov 1;49(5):1941-1961. doi: 10.1042/BST20200604. Biochem Soc Trans. 2021. PMID: 34643236 Free PMC article. Review.
Cited by
-
Carbon monoxide induces cardiac arrhythmia via induction of the late Na+ current.Am J Respir Crit Care Med. 2012 Oct 1;186(7):648-56. doi: 10.1164/rccm.201204-0688OC. Epub 2012 Jul 19. Am J Respir Crit Care Med. 2012. PMID: 22822026 Free PMC article.
-
CO-Releasing Molecules Have Nonheme Targets in Bacteria: Transcriptomic, Mathematical Modeling and Biochemical Analyses of CORM-3 [Ru(CO)3Cl(glycinate)] Actions on a Heme-Deficient Mutant of Escherichia coli.Antioxid Redox Signal. 2015 Jul 10;23(2):148-62. doi: 10.1089/ars.2014.6151. Epub 2015 Apr 28. Antioxid Redox Signal. 2015. PMID: 25811604 Free PMC article.
-
Carbon monoxide in lung cell physiology and disease.Am J Physiol Cell Physiol. 2018 Feb 1;314(2):C211-C227. doi: 10.1152/ajpcell.00022.2017. Epub 2017 Nov 8. Am J Physiol Cell Physiol. 2018. PMID: 29118026 Free PMC article. Review.
-
Air Pollution and Cardiac Arrhythmias: From Epidemiological and Clinical Evidences to Cellular Electrophysiological Mechanisms.Front Cardiovasc Med. 2021 Oct 28;8:736151. doi: 10.3389/fcvm.2021.736151. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 34778399 Free PMC article. Review.
-
Amantadine Combines Astroglial System Xc- Activation with Glutamate/NMDA Receptor Inhibition.Biomolecules. 2019 May 17;9(5):191. doi: 10.3390/biom9050191. Biomolecules. 2019. PMID: 31108896 Free PMC article.
References
-
- Wu L., Wang R. (2005) Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol. Rev. 57, 585–630 - PubMed
-
- Kim H. P., Ryter S. W., Choi A. M. (2006) CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 46, 411–449 - PubMed
-
- Ryter S. W., Alam J., Choi A. M. (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 86, 583–650 - PubMed
-
- Lakkisto P., Palojoki E., Bäcklund T., Saraste A., Tikkanen I., Voipio-Pulkki L. M., Pulkki K. (2002) Expression of heme oxygenase-1 in response to myocardial infarction in rats. J. Mol. Cell Cardiol. 34, 1357–1365 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
