

Mixed triple: allied viruses in unique recent isolates of highly virulent type 2 bovine viral diarrhea virus detected by deep sequencing
- PMID: 24719408
- PMCID: PMC4054349
- DOI: 10.1128/JVI.00620-14
Mixed triple: allied viruses in unique recent isolates of highly virulent type 2 bovine viral diarrhea virus detected by deep sequencing
Abstract
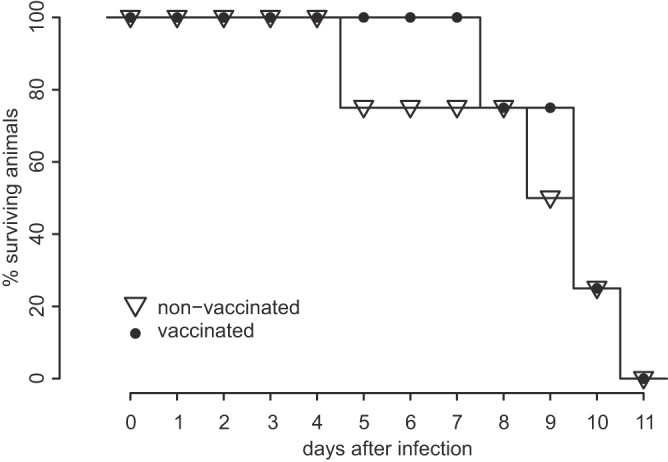
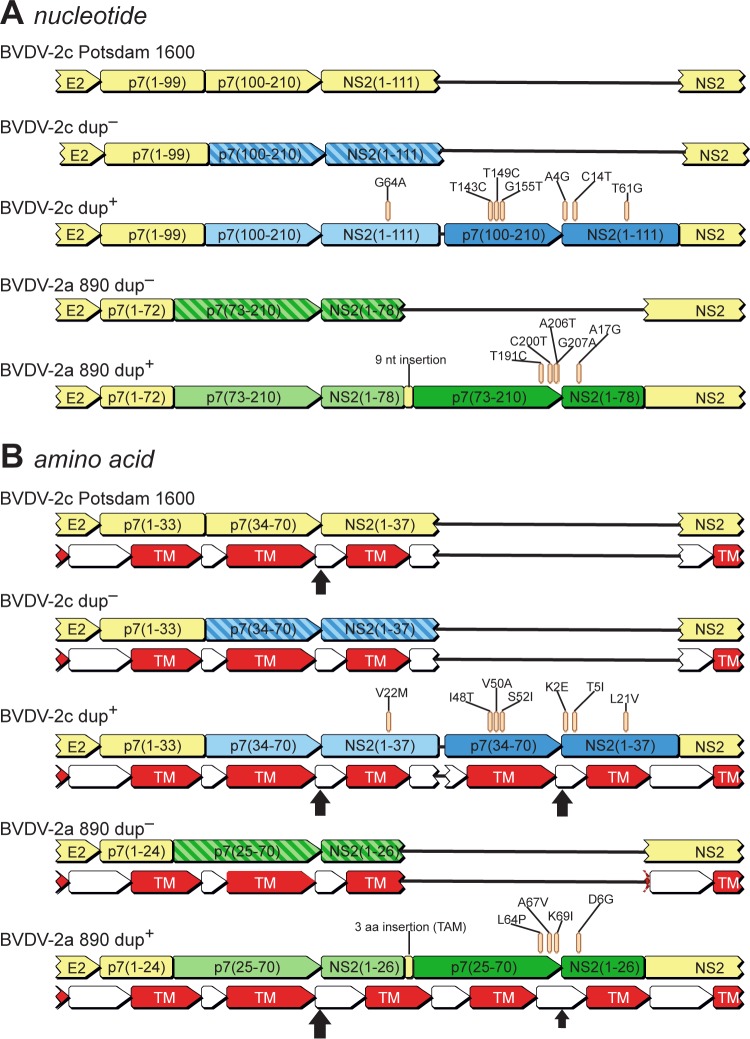
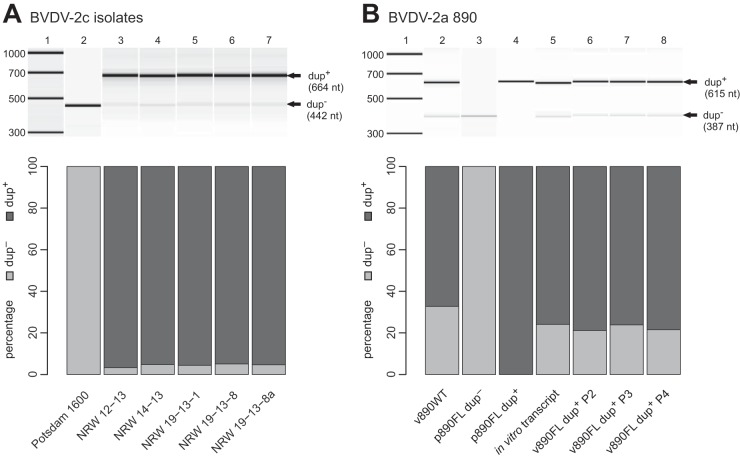
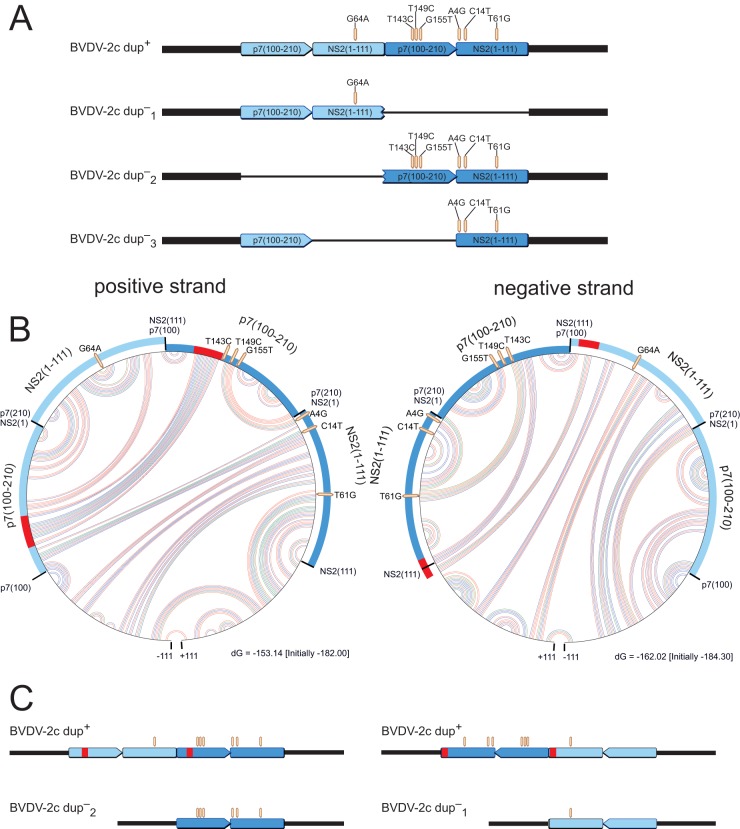
In February 2013, very severe acute clinical symptoms were observed in calves, heifers, and dairy cattle in several farms in North Rhine Westphalia and Lower Saxony, Germany. Deep sequencing revealed the coexistence of three distinct genome variants within recent highly virulent bovine viral diarrhea virus type 2 (BVDV-2) isolates. While the major portion (ca. 95%) of the population harbored a duplication of a 222-nucleotide (nt) segment within the p7-NS2-encoding region, the minority reflected the standard structure of a BVDV-2 genome. Additionally, unusual mutations were found in both variants, within the highly conserved p7 protein and close to the p7-NS2 cleavage site. Using a reverse genetic system with a BVDV-2a strain harboring a similar duplication, it could be demonstrated that during replication, genomes without duplication are generated de novo from genomes with duplication. The major variant with duplication is compulsorily escorted by the minor variant without duplication. RNA secondary structure prediction allowed the analysis of the unique but stable mixture of three BVDV variants and also provided the explanation for their generation. Finally, our results suggest that the variant with duplication plays the major role in the highly virulent phenotype.
Importance: This study emphasizes the importance of full-genome deep sequencing in combination with manual in-depth data analysis for the investigation of viruses in basic research and diagnostics. Here we investigated recent highly virulent bovine viral diarrhea virus isolates from a 2013 series of outbreaks. We discovered a unique special feature of the viral genome, an unstable duplication of 222 nucleotides which is eventually deleted by viral polymerase activity, leading to an unexpectedly mixed population of viral genomes for all investigated isolates. Our study is of high importance to the field because we demonstrate that these insertion/deletion events allow another level of genome plasticity of plus-strand RNA viruses, in addition to the well-known polymerase-induced single nucleotide variations which are generally considered the main basis for viral adaptation and evolution.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Nonhomologous RNA recombination in bovine viral diarrhea virus: molecular characterization of a variety of subgenomic RNAs isolated during an outbreak of fatal mucosal disease.J Virol. 1999 Jul;73(7):5646-53. doi: 10.1128/JVI.73.7.5646-5653.1999. J Virol. 1999. PMID: 10364314 Free PMC article.
-
Prevalence of genotypes 1 and 2 of bovine viral diarrhea virus in Lower Saxony, Germany.Virus Res. 2001 Jul;76(1):31-42. doi: 10.1016/s0168-1702(01)00244-1. Virus Res. 2001. PMID: 11376844
-
Genetic change in the open reading frame of bovine viral diarrhea virus is introduced more rapidly during the establishment of a single persistent infection than from multiple acute infections.Virus Res. 2011 Jun;158(1-2):140-5. doi: 10.1016/j.virusres.2011.03.024. Epub 2011 Apr 4. Virus Res. 2011. PMID: 21470568
-
Characterisation of a type 2 bovine viral diarrhoea virus isolated from cattle in the UK.Vet Microbiol. 2004 Aug 19;102(1-2):19-24. doi: 10.1016/j.vetmic.2004.05.005. Vet Microbiol. 2004. PMID: 15288923
-
Genetic characterization of a noncytopathic bovine viral diarrhea virus 2b isolated from cattle in China.Virus Genes. 2014 Oct;49(2):339-41. doi: 10.1007/s11262-014-1067-7. Epub 2014 May 9. Virus Genes. 2014. PMID: 24811746
Cited by
-
Genetic Diversity of Bovine Viral Diarrhea Virus Infection in Goats in Southwestern China.J Vet Med. 2018 Nov 18;2018:8274397. doi: 10.1155/2018/8274397. eCollection 2018. J Vet Med. 2018. PMID: 30581873 Free PMC article.
-
Identification of a new bovine viral diarrhea virus subtype in the Republic of Korea.BMC Vet Res. 2018 Aug 7;14(1):233. doi: 10.1186/s12917-018-1555-4. BMC Vet Res. 2018. PMID: 30086756 Free PMC article.
-
Genetic and antigenic characterization of bovine viral diarrhea viruses isolated from cattle in Hokkaido, Japan.J Vet Med Sci. 2016 Jan;78(1):61-70. doi: 10.1292/jvms.15-0186. Epub 2015 Sep 21. J Vet Med Sci. 2016. PMID: 26400674 Free PMC article.
-
Use of Genomic Tools to Improve Cattle Health in the Context of Infectious Diseases.Front Genet. 2016 Mar 7;7:30. doi: 10.3389/fgene.2016.00030. eCollection 2016. Front Genet. 2016. PMID: 27014337 Free PMC article. Review.
-
A newly developed BVDV-1 RT-qPCR Taqman assay based on Italian isolates: evaluation as a diagnostic tool.Folia Microbiol (Praha). 2017 Jul;62(4):279-286. doi: 10.1007/s12223-017-0497-8. Epub 2017 Jan 26. Folia Microbiol (Praha). 2017. PMID: 28127668
References
-
- Schirrmeier H. 2013. Auftreten von akuten Infektionen mit BVDV-2c in Deutschland. Mitt. Ganzen Nord. 2:17–21
-
- ProMED-Mail. 26 March 2013. Bovine viral diarrhea—Netherlands: (GE), BVDV type II. Archive number 20130326.1603817. http://www.promedmail.org/
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
