

Regulation of Potassium Homeostasis
- PMID: 24721891
- PMCID: PMC4455213
- DOI: 10.2215/CJN.08580813
Regulation of Potassium Homeostasis
Abstract
Potassium is the most abundant cation in the intracellular fluid, and maintaining the proper distribution of potassium across the cell membrane is critical for normal cell function. Long-term maintenance of potassium homeostasis is achieved by alterations in renal excretion of potassium in response to variations in intake. Understanding the mechanism and regulatory influences governing the internal distribution and renal clearance of potassium under normal circumstances can provide a framework for approaching disorders of potassium commonly encountered in clinical practice. This paper reviews key aspects of the normal regulation of potassium metabolism and is designed to serve as a readily accessible review for the well informed clinician as well as a resource for teaching trainees and medical students.
Keywords: aldosterone; collecting duct; potassium; renal physiology; tubular transport.
Copyright © 2015 by the American Society of Nephrology.
Figures
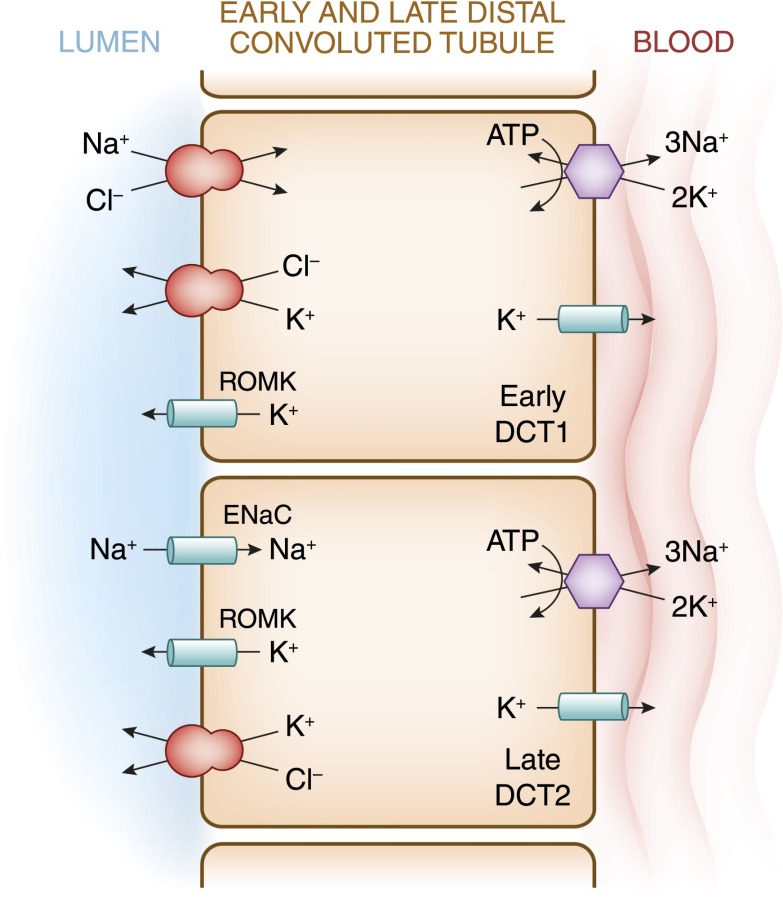
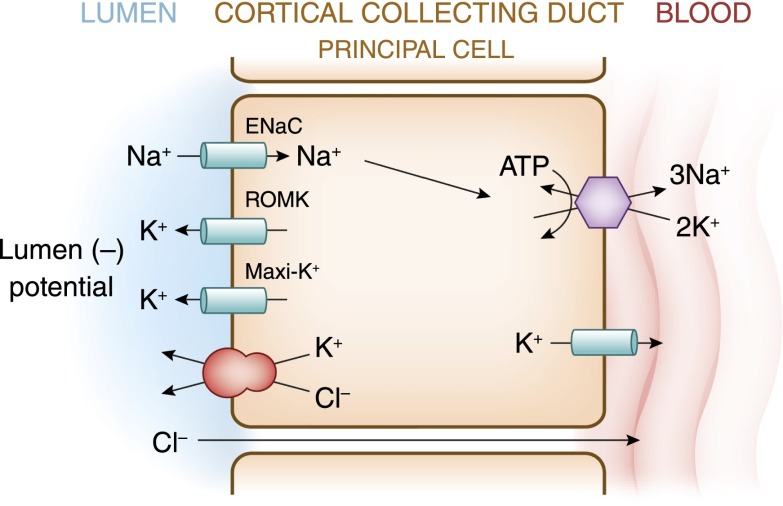
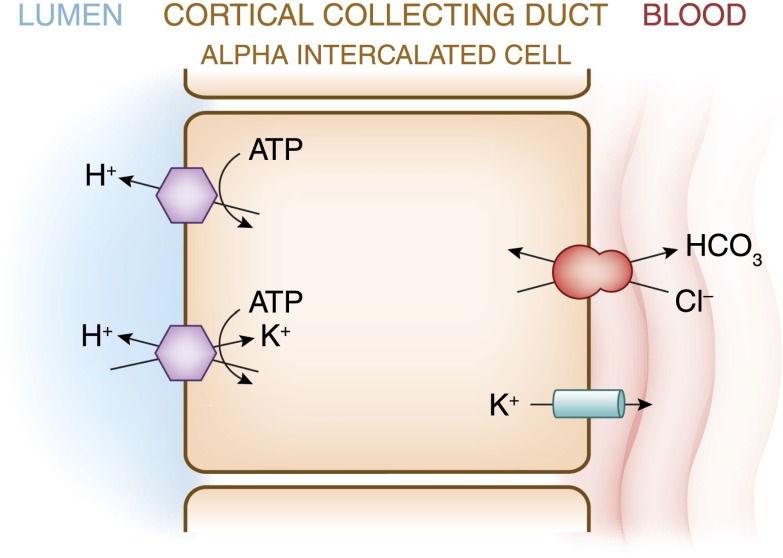
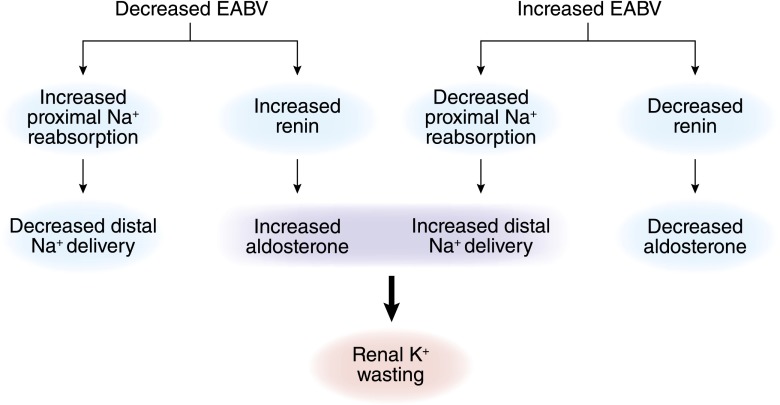
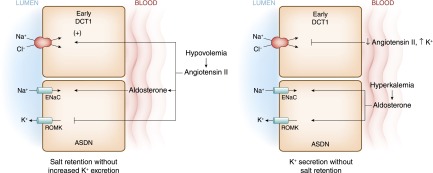
References
-
- Palmer BF: A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis 56: 387–393, 2010 - PubMed
-
- Foley K, Boguslavsky S, Klip A: Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 50: 3048–3061, 2011 - PubMed
-
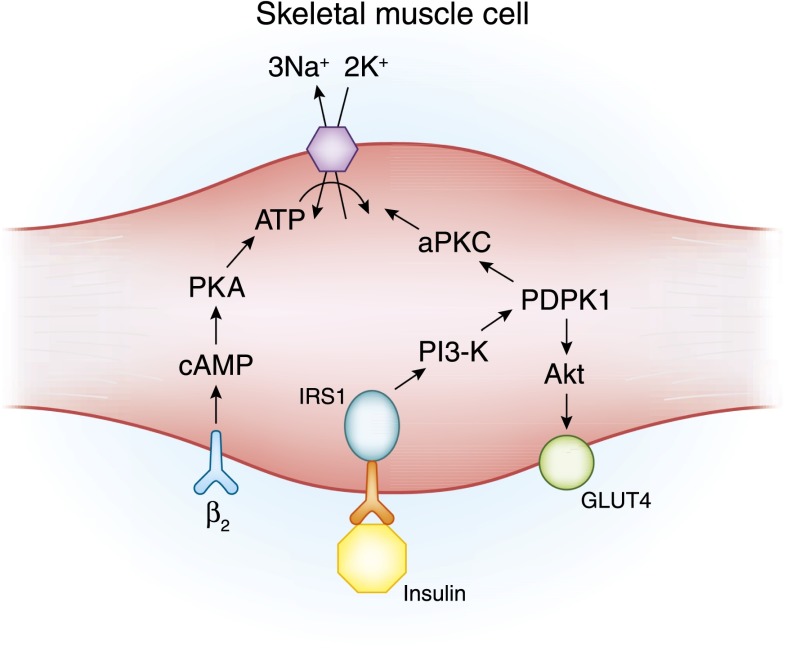
- Ho K: A critically swift response: Insulin-stimulated potassium and glucose transport in skeletal muscle. Clin J Am Soc Nephrol 6: 1513–1516, 2011 - PubMed
-
- Alvestrand A, Wahren J, Smith D, DeFronzo RA: Insulin-mediated potassium uptake is normal in uremic and healthy subjects. Am J Physiol 246: E174–E180, 1984 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
