

Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia
- PMID: 24724051
- PMCID: PMC3971203
- DOI: 10.3389/fonc.2014.00054
Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia
Abstract
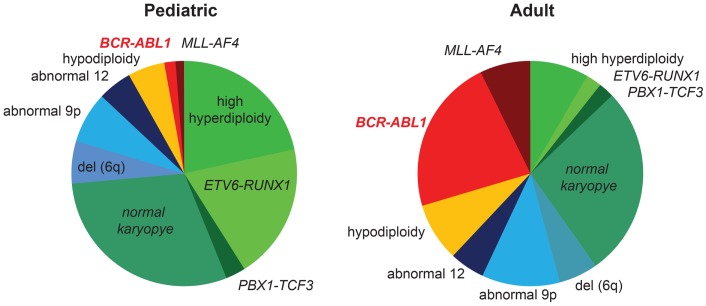
The t(9;22)(q34;q11) or Philadelphia chromosome creates a BCR-ABL1 fusion gene encoding for a chimeric BCR-ABL1 protein. It is present in 3-4% of pediatric acute lymphoblastic leukemia (Ph(+) ALL), and about 25% of adult ALL cases. Prior to the advent of tyrosine kinase inhibitors (TKI), Ph(+) ALL was associated with a very poor prognosis despite the use of intensive chemotherapy and frequently hematopoietic stem-cell transplantation (HSCT) in first remission. The development of TKIs revolutionized the therapy of Ph(+) ALL. Addition of the first generation ABL1 class TKI imatinib to intensive chemotherapy dramatically increased the survival for children with Ph(+) ALL and established that many patients can be cured without HSCT. In parallel, the mechanistic understanding of Ph(+) ALL expanded exponentially through careful mapping of pathways downstream of BCR-ABL1, the discovery of mutations in master regulators of B-cell development such as IKZF1 (Ikaros), PAX5, and early B-cell factor (EBF), the recognition of the complex clonal architecture of Ph(+) ALL, and the delineation of genomic, epigenetic, and signaling abnormalities contributing to relapse and resistance. Still, many important basic and clinical questions remain unanswered. Current clinical trials are testing second generation TKIs in patients with newly diagnosed Ph(+) ALL. Neither the optimal duration of therapy nor the optimal chemotherapy backbone are currently defined. The role of HSCT in first remission and post-transplant TKI therapy also require further study. In addition, it will be crucial to continue to dig deeper into understanding Ph(+) ALL at a mechanistic level, and translate findings into complementary targeted approaches. Expanding targeted therapies hold great promise to decrease toxicity and improve survival in this high-risk disease, which provides a paradigm for how targeted therapies can be incorporated into treatment of other high-risk leukemias.
Keywords: BCR–ABL1; acute lymphoblastic leukemia; chemotherapy; hematopoietic stem-cell transplantation; tyrosine kinase inhibition.
Figures





References
-
- Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. J Clin Oncol (2009) 27(31):5175–8110.1200/JCO.2008.21.2514 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous