

BAYSIC: a Bayesian method for combining sets of genome variants with improved specificity and sensitivity
- PMID: 24725768
- PMCID: PMC3999887
- DOI: 10.1186/1471-2105-15-104
BAYSIC: a Bayesian method for combining sets of genome variants with improved specificity and sensitivity
Abstract
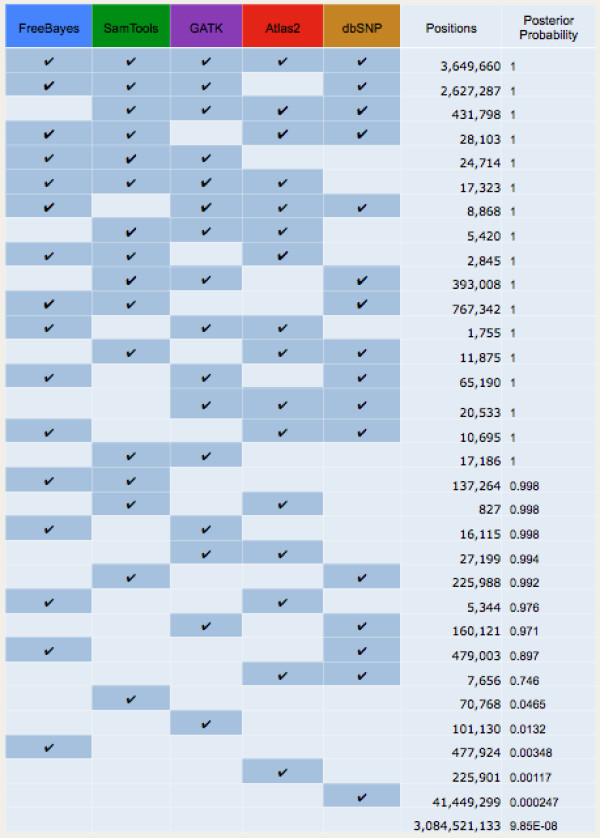
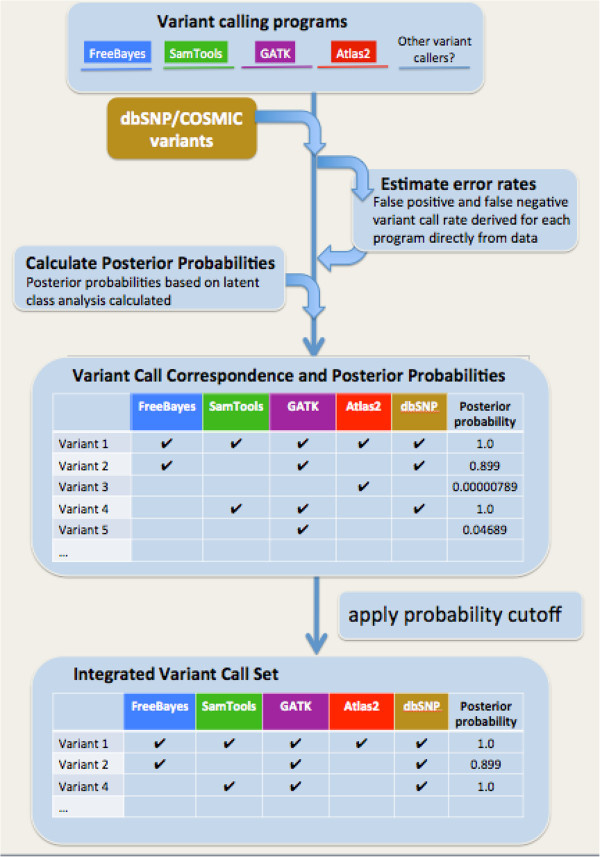
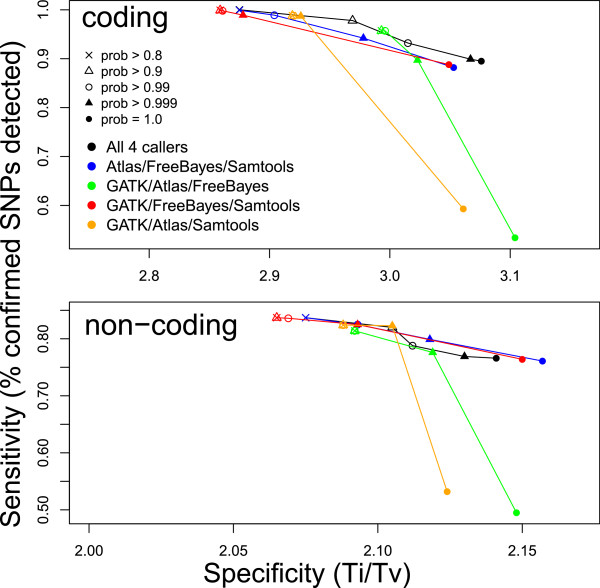
Background: Accurate genomic variant detection is an essential step in gleaning medically useful information from genome data. However, low concordance among variant-calling methods reduces confidence in the clinical validity of whole genome and exome sequence data, and confounds downstream analysis for applications in genome medicine.Here we describe BAYSIC (BAYeSian Integrated Caller), which combines SNP variant calls produced by different methods (e.g. GATK, FreeBayes, Atlas, SamTools, etc.) into a more accurate set of variant calls. BAYSIC differs from majority voting, consensus or other ad hoc intersection-based schemes for combining sets of genome variant calls. Unlike other classification methods, the underlying BAYSIC model does not require training using a "gold standard" of true positives. Rather, with each new dataset, BAYSIC performs an unsupervised, fully Bayesian latent class analysis to estimate false positive and false negative error rates for each input method. The user specifies a posterior probability threshold according to the user's tolerance for false positive and false negative errors; lowering the posterior probability threshold allows the user to trade specificity for sensitivity while raising the threshold increases specificity in exchange for sensitivity.
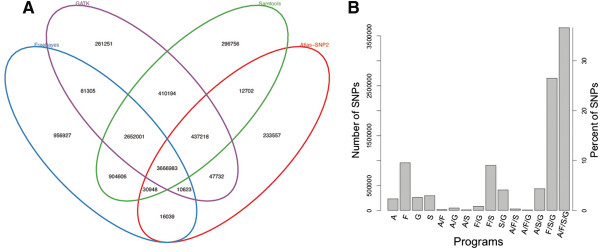
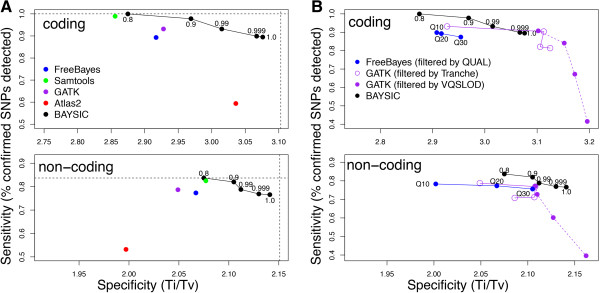
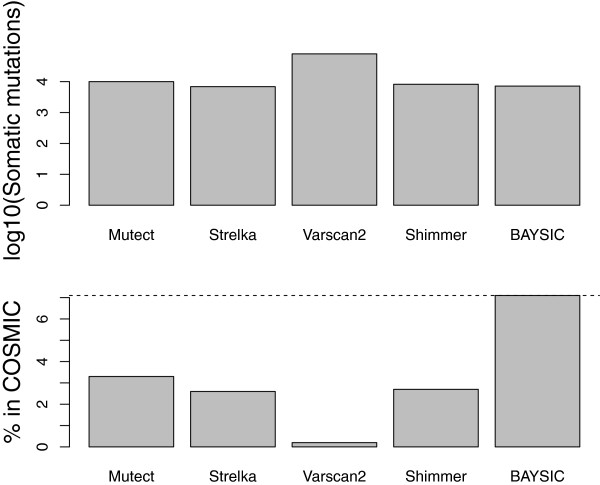
Results: We assessed the performance of BAYSIC in comparison to other variant detection methods using ten low coverage (~5X) samples from The 1000 Genomes Project, a tumor/normal exome pair (40X), and exome sequences (40X) from positive control samples previously identified to contain clinically relevant SNPs. We demonstrated BAYSIC's superior variant-calling accuracy, both for somatic mutation detection and germline variant detection.
Conclusions: BAYSIC provides a method for combining sets of SNP variant calls produced by different variant calling programs. The integrated set of SNP variant calls produced by BAYSIC improves the sensitivity and specificity of the variant calls used as input. In addition to combining sets of germline variants, BAYSIC can also be used to combine sets of somatic mutations detected in the context of tumor/normal sequencing experiments.
Figures
References
-
- Martin ADG, Kamm T, Ordowski M, Przybocki M. The DET curve in assessment of detection task performance. Proc Eurospeech. 1899–1903;1997:4.
-
- Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, Merker JD, Goldfeder RL, Enns GM, David SP, Pakdaman N, Ormond KE, Caleshu C, Kingham K, Klein TE, Whirl-Carrillo M, Sakamoto K, Wheeler MT, Butte AJ, Ford JM, Boxer L, Ioannidis JP, Yeung AC, Altman RB, Assimes TL, Snyder M, Ashley EA Quertermous T. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311(10):1035–1045. doi: 10.1001/jama.2014.1717. - DOI - PMC - PubMed
-
- Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, Buhay CJ, Kovar CL, Wang M, Hawes AC, Reid JG, Eng C, Muzny DM, Gibbs RA. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med. 2013;5(6):57. - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
