

Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance
- PMID: 24727571
- PMCID: PMC3999722
- DOI: 10.1093/brain/awu060
Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance
Abstract
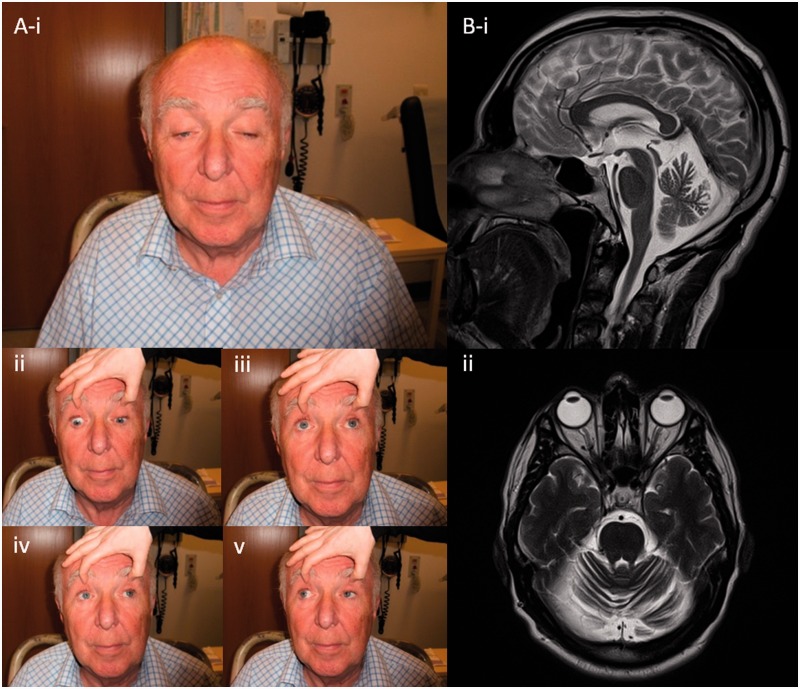
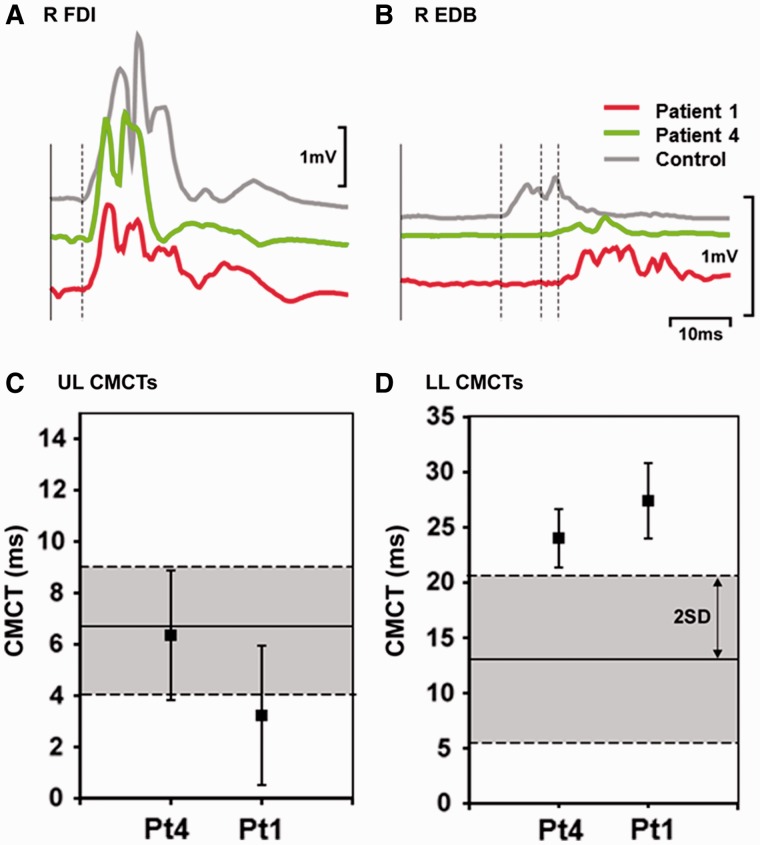
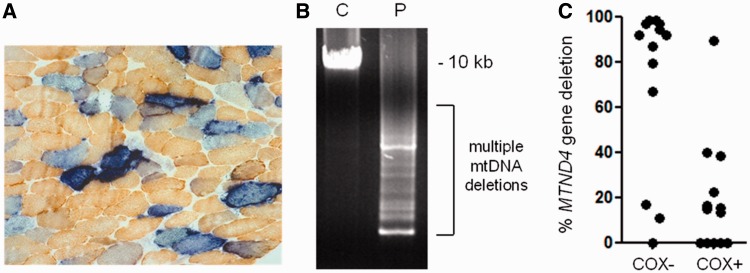
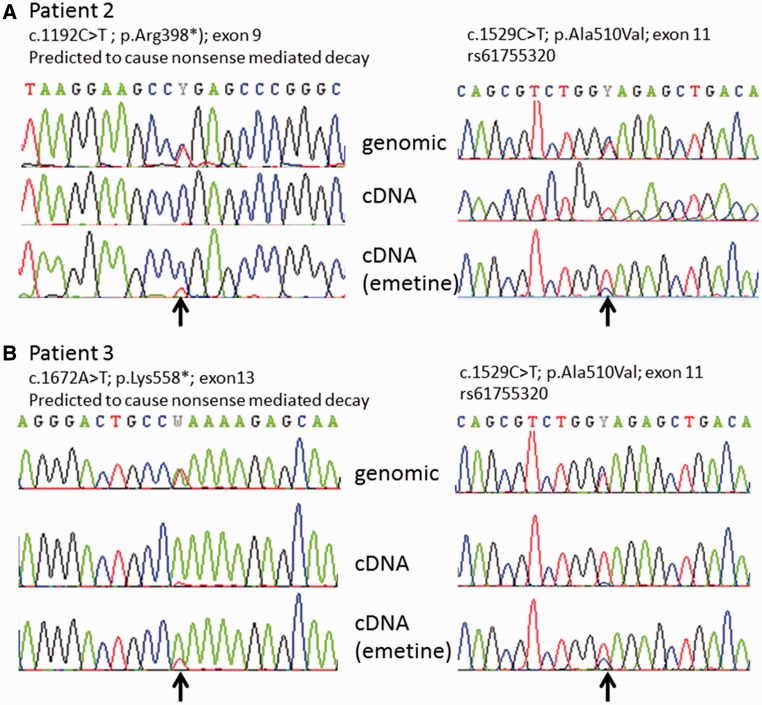
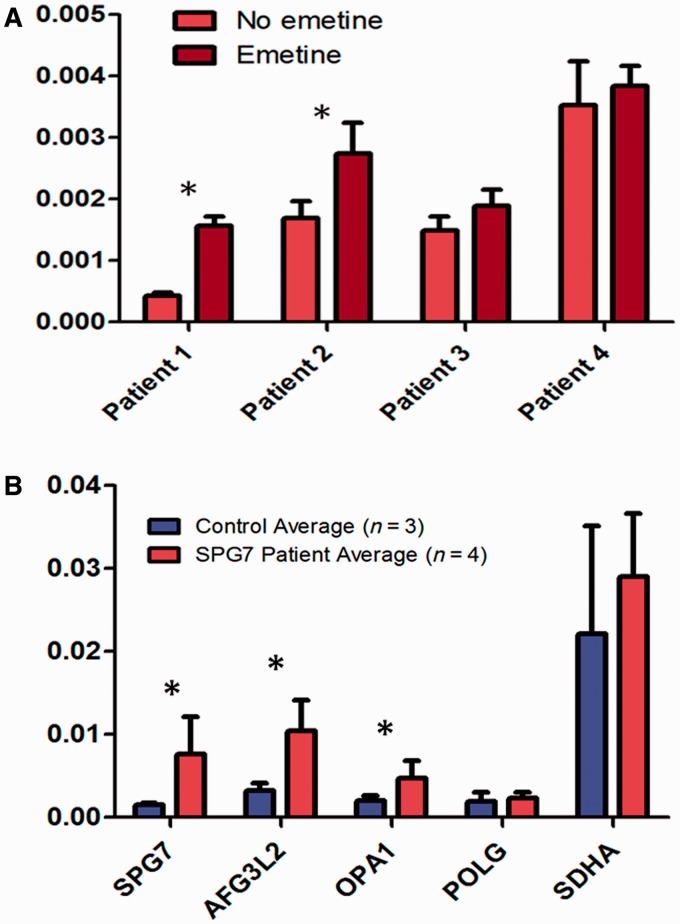
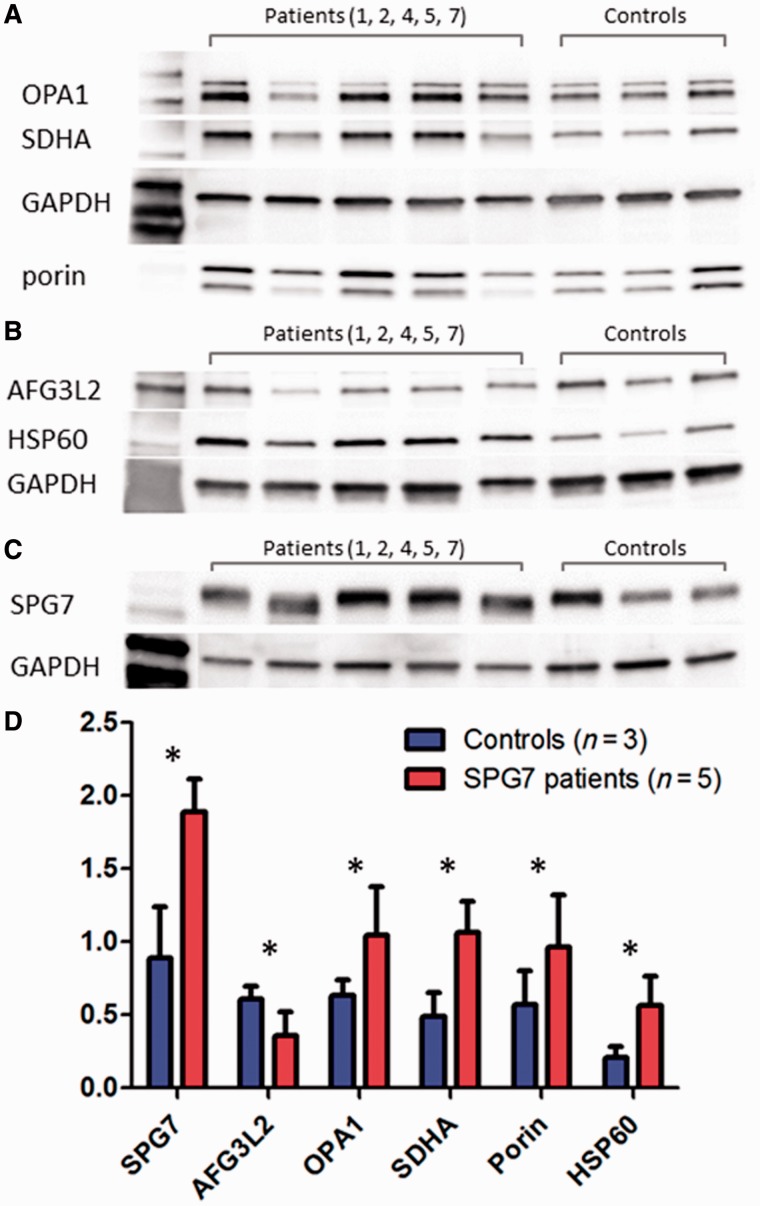
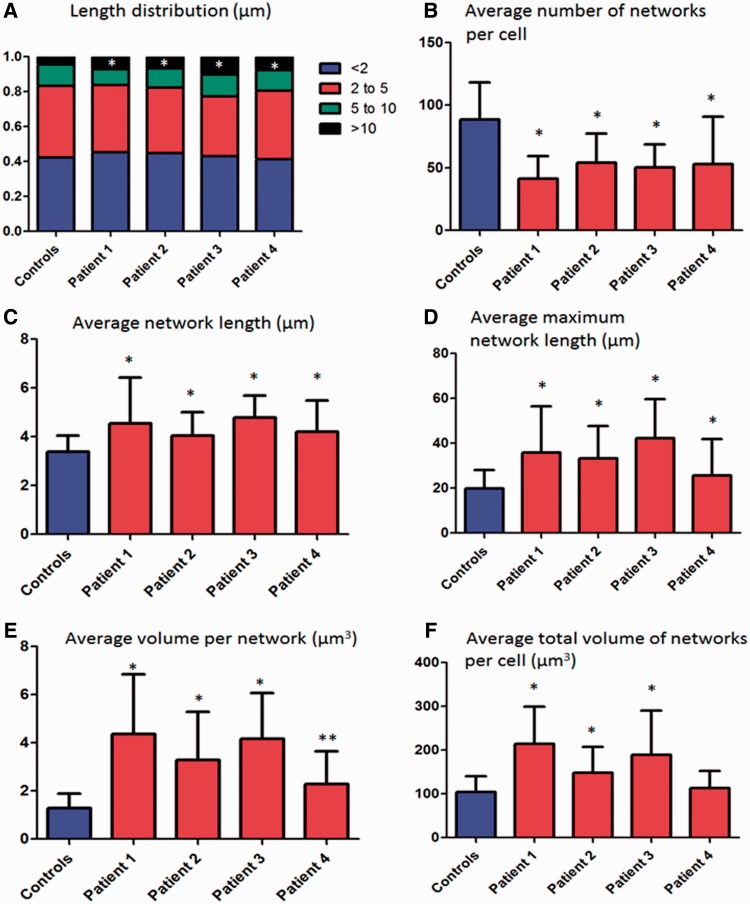
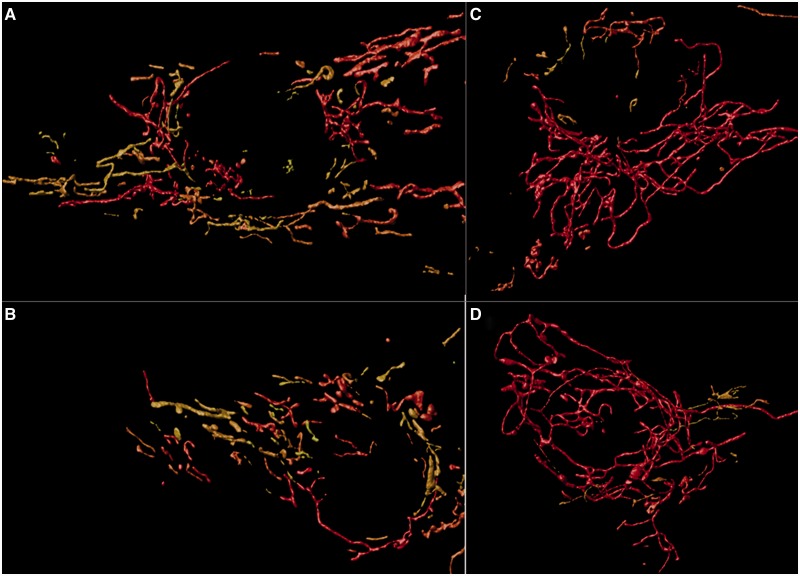
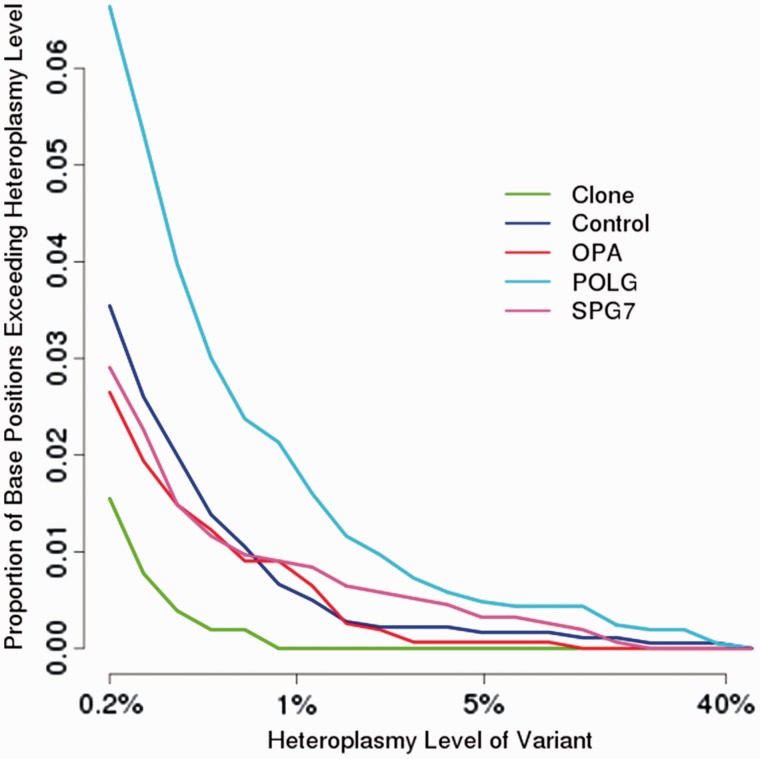
Despite being a canonical presenting feature of mitochondrial disease, the genetic basis of progressive external ophthalmoplegia remains unknown in a large proportion of patients. Here we show that mutations in SPG7 are a novel cause of progressive external ophthalmoplegia associated with multiple mitochondrial DNA deletions. After excluding known causes, whole exome sequencing, targeted Sanger sequencing and multiplex ligation-dependent probe amplification analysis were used to study 68 adult patients with progressive external ophthalmoplegia either with or without multiple mitochondrial DNA deletions in skeletal muscle. Nine patients (eight probands) were found to carry compound heterozygous SPG7 mutations, including three novel mutations: two missense mutations c.2221G>A; p.(Glu741Lys), c.2224G>A; p.(Asp742Asn), a truncating mutation c.861dupT; p.Asn288*, and seven previously reported mutations. We identified a further six patients with single heterozygous mutations in SPG7, including two further novel mutations: c.184-3C>T (predicted to remove a splice site before exon 2) and c.1067C>T; p.(Thr356Met). The clinical phenotype typically developed in mid-adult life with either progressive external ophthalmoplegia/ptosis and spastic ataxia, or a progressive ataxic disorder. Dysphagia and proximal myopathy were common, but urinary symptoms were rare, despite the spasticity. Functional studies included transcript analysis, proteomics, mitochondrial network analysis, single fibre mitochondrial DNA analysis and deep re-sequencing of mitochondrial DNA. SPG7 mutations caused increased mitochondrial biogenesis in patient muscle, and mitochondrial fusion in patient fibroblasts associated with the clonal expansion of mitochondrial DNA mutations. In conclusion, the SPG7 gene should be screened in patients in whom a disorder of mitochondrial DNA maintenance is suspected when spastic ataxia is prominent. The complex neurological phenotype is likely a result of the clonal expansion of secondary mitochondrial DNA mutations modulating the phenotype, driven by compensatory mitochondrial biogenesis.
Keywords: SPG7; chronic progressive external ophthalmoplegia; hereditary spastic paraplegia; mtDNA maintenance; paraplegin.
Figures
References
-
- Arnoldi A, Tonelli A, Crippa F, Villani G, Pacelli C, Sironi M, et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat. 2008;29:522–31. - PubMed
-
- Blakely E, He L, Gardner JL, Hudson G, Walter J, Hughes I, et al. Novel mutations in the TK2 gene associated with fatal mitochondrial DNA depletion myopathy. Neuromuscul Disord. 2008;18:557–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- G1002570/MRC_/Medical Research Council/United Kingdom
- G0701386/MRC_/Medical Research Council/United Kingdom
- MR/K000608/1/MRC_/Medical Research Council/United Kingdom
- G0800674/MRC_/Medical Research Council/United Kingdom
- MR/K006312/1/MRC_/Medical Research Council/United Kingdom
- NIHR-HCS-D12-03-04/DH_/Department of Health/United Kingdom
- 084980/Z/08/Z/WT_/Wellcome Trust/United Kingdom
- G0601943/DH_/Department of Health/United Kingdom
- G0601943/MRC_/Medical Research Council/United Kingdom
- 096919Z/11/Z/WT_/Wellcome Trust/United Kingdom
- 101876/WT_/Wellcome Trust/United Kingdom
- G0800470/MRC_/Medical Research Council/United Kingdom
- 096919/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
