

Multiscale representation of genomic signals
- PMID: 24727652
- PMCID: PMC4040162
- DOI: 10.1038/nmeth.2924
Multiscale representation of genomic signals
Abstract
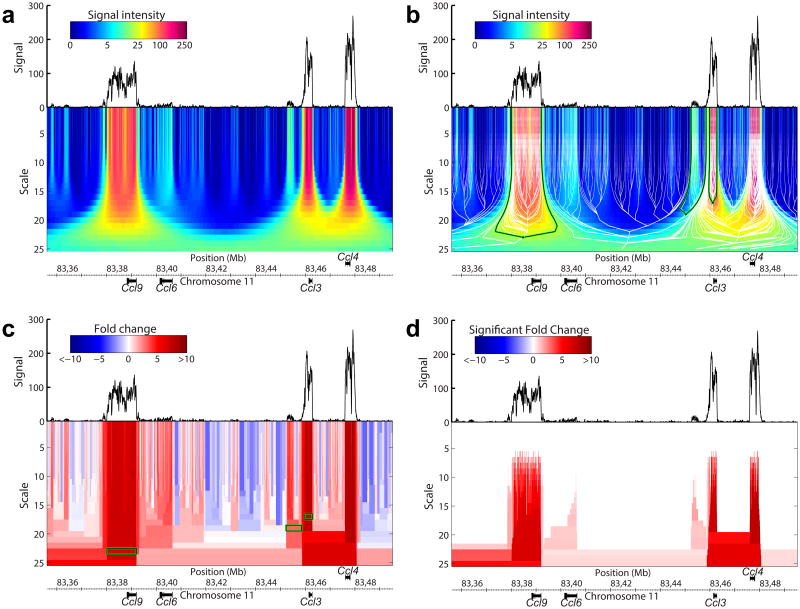
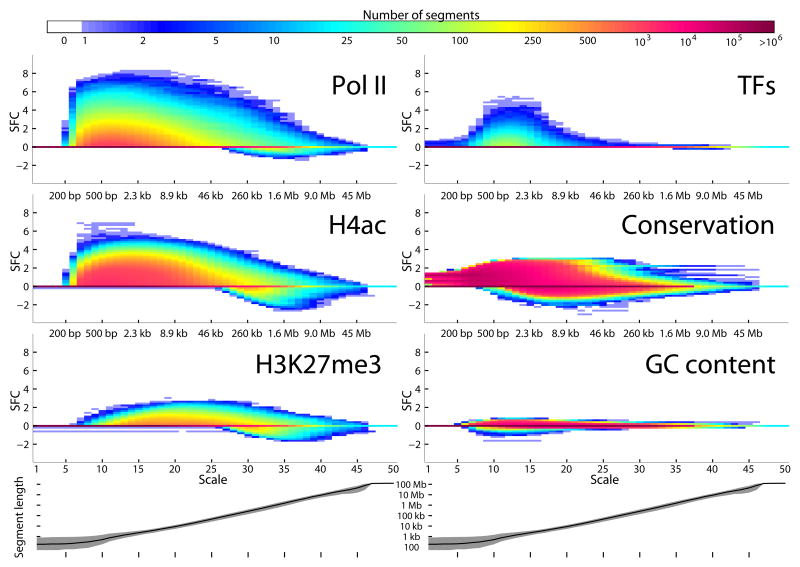
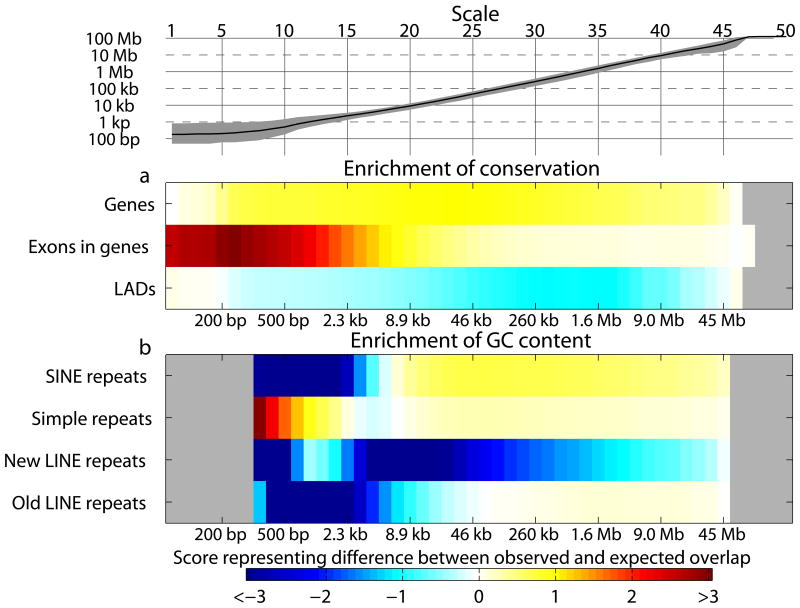
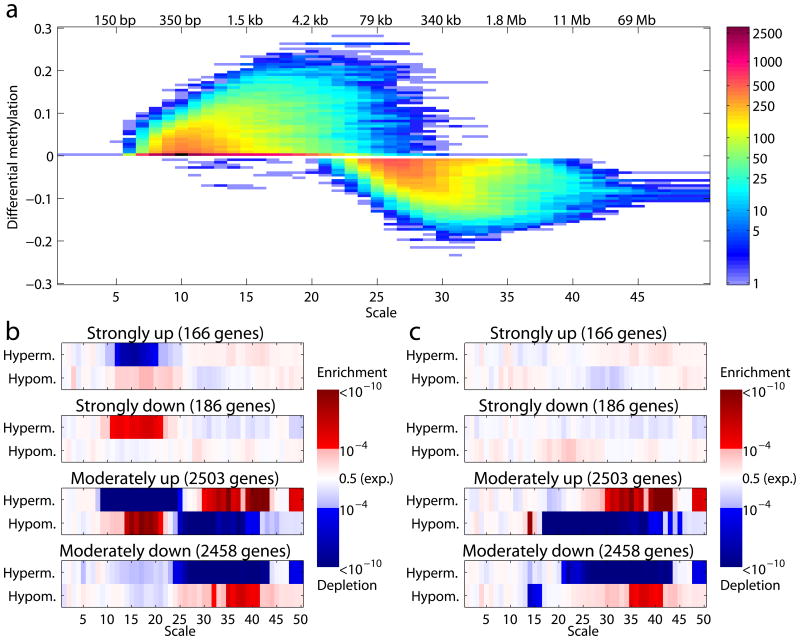
Genomic information is encoded on a wide range of distance scales, ranging from tens of bases to megabases. We developed a multiscale framework to analyze and visualize the information content of genomic signals. Different types of signals, such as G+C content or DNA methylation, are characterized by distinct patterns of signal enrichment or depletion across scales spanning several orders of magnitude. These patterns are associated with a variety of genomic annotations. By integrating the information across all scales, we demonstrated improved prediction of gene expression from polymerase II chromatin immunoprecipitation sequencing (ChIP-seq) measurements, and we observed that gene expression differences in colorectal cancer are related to methylation patterns that extend beyond the single-gene scale. Our software is available at https://github.com/tknijnen/msr/.
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
