

Classification and evolution of type II CRISPR-Cas systems
- PMID: 24728998
- PMCID: PMC4041416
- DOI: 10.1093/nar/gku241
Classification and evolution of type II CRISPR-Cas systems
Abstract
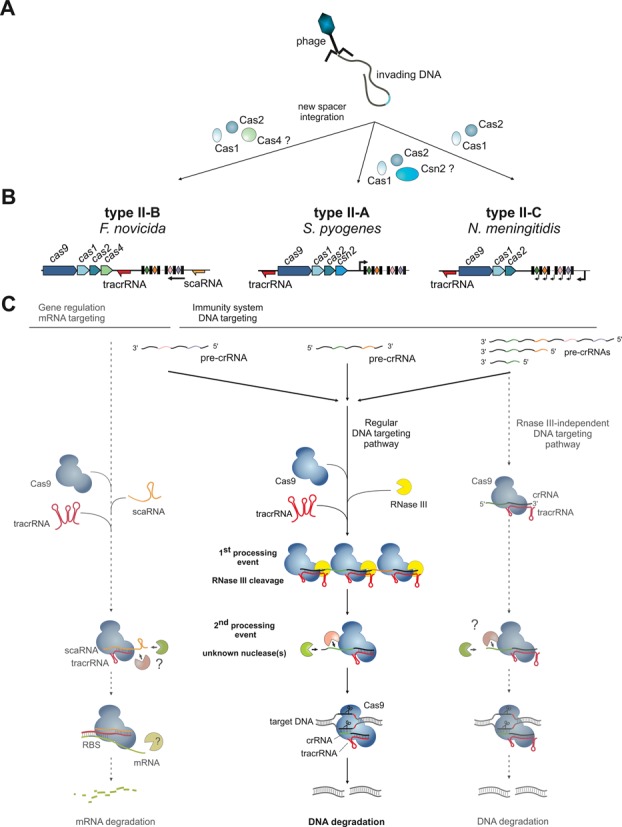
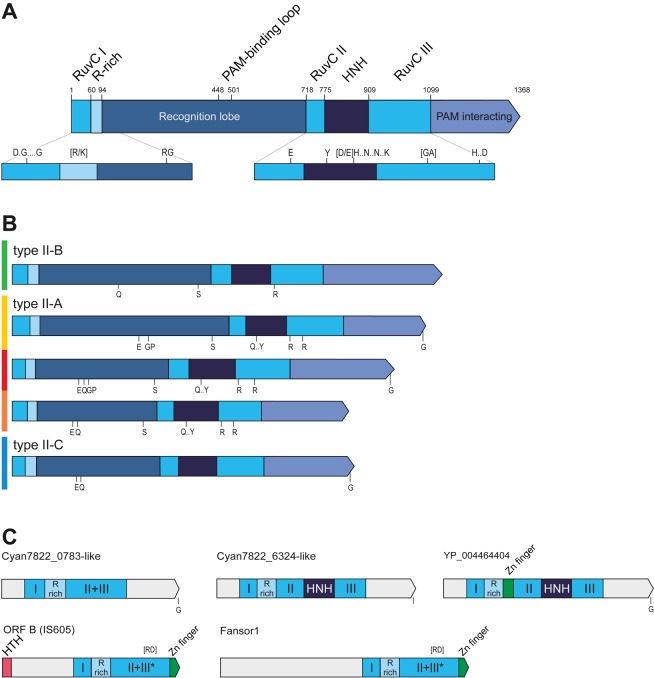
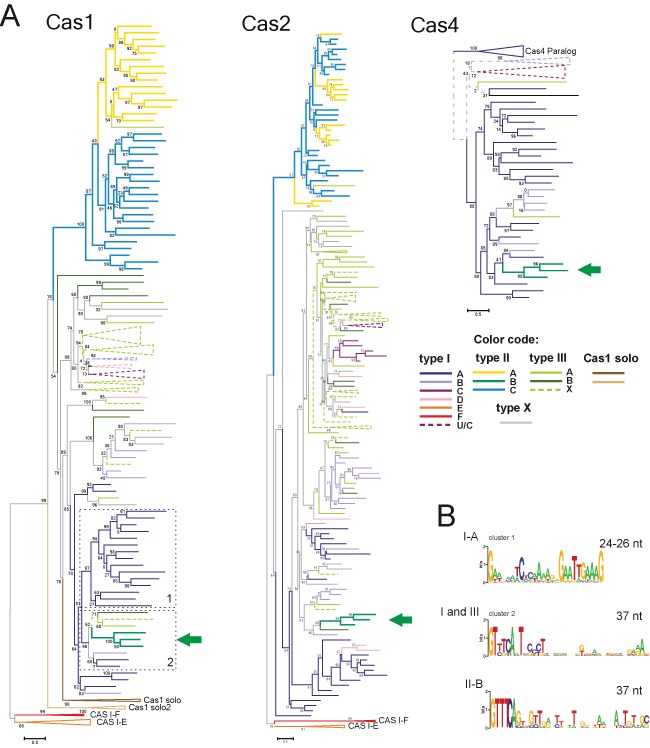
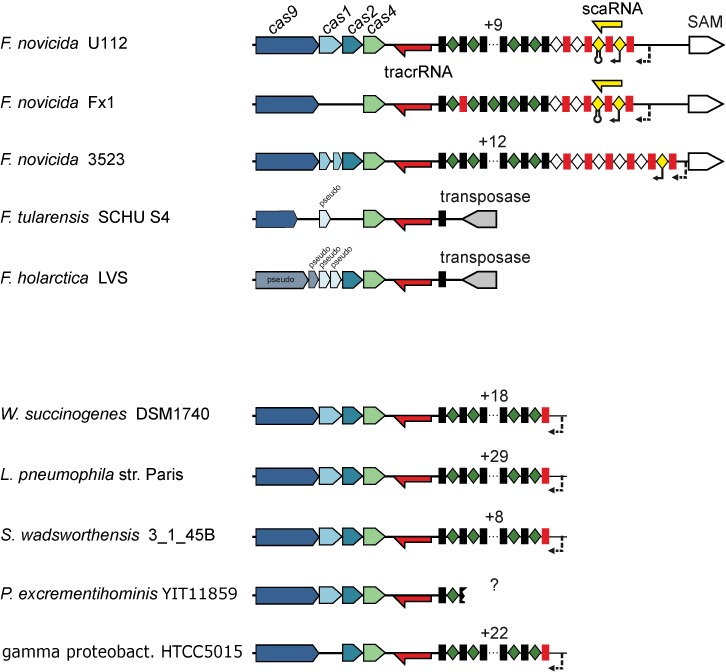
The CRISPR-Cas systems of archaeal and bacterial adaptive immunity are classified into three types that differ by the repertoires of CRISPR-associated (cas) genes, the organization of cas operons and the structure of repeats in the CRISPR arrays. The simplest among the CRISPR-Cas systems is type II in which the endonuclease activities required for the interference with foreign deoxyribonucleic acid (DNA) are concentrated in a single multidomain protein, Cas9, and are guided by a co-processed dual-tracrRNA:crRNA molecule. This compact enzymatic machinery and readily programmable site-specific DNA targeting make type II systems top candidates for a new generation of powerful tools for genomic engineering. Here we report an updated census of CRISPR-Cas systems in bacterial and archaeal genomes. Type II systems are the rarest, missing in archaea, and represented in ∼ 5% of bacterial genomes, with an over-representation among pathogens and commensals. Phylogenomic analysis suggests that at least three cas genes, cas1, cas2 and cas4, and the CRISPR repeats of the type II-B system were acquired via recombination with a type I CRISPR-Cas locus. Distant homologs of Cas9 were identified among proteins encoded by diverse transposons, suggesting that type II CRISPR-Cas evolved via recombination of mobile nuclease genes with type I loci.
Published by Oxford University Press on behalf of Nucleic Acids Research 2014. This work is written by (a) US Government employee(s) and is in the public domain in the US.
Figures


References
-
- Barrangou R., Horvath P. CRISPR: new horizons in phage resistance and strain identification. Annu. Rev. Food Sci. Technol. 2012;3:143–162. - PubMed
-
- Wiedenheft B., Sternberg S.H., Doudna J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. - PubMed
-
- van der Oost J., Jore M.M., Westra E.R., Lundgren M., Brouns S.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009;34:401–407. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
