

Designing and evaluating the MULTICOM protein local and global model quality prediction methods in the CASP10 experiment
- PMID: 24731387
- PMCID: PMC3996498
- DOI: 10.1186/1472-6807-14-13
Designing and evaluating the MULTICOM protein local and global model quality prediction methods in the CASP10 experiment
Abstract
Background: Protein model quality assessment is an essential component of generating and using protein structural models. During the Tenth Critical Assessment of Techniques for Protein Structure Prediction (CASP10), we developed and tested four automated methods (MULTICOM-REFINE, MULTICOM-CLUSTER, MULTICOM-NOVEL, and MULTICOM-CONSTRUCT) that predicted both local and global quality of protein structural models.
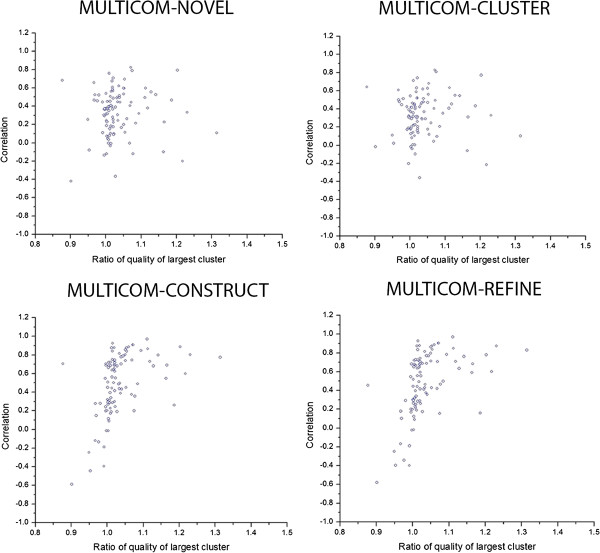
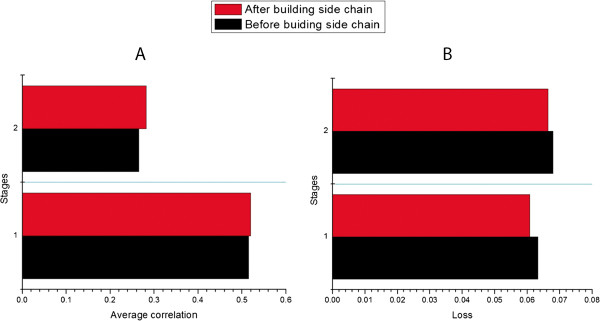
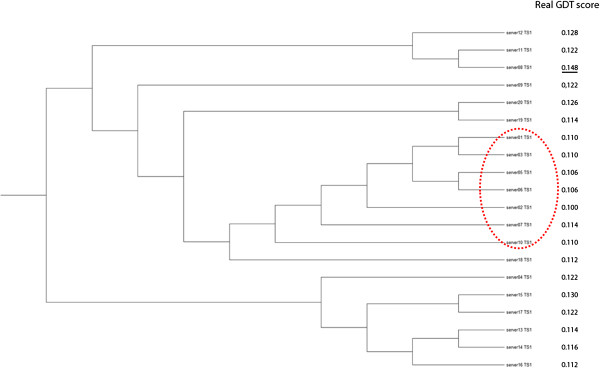
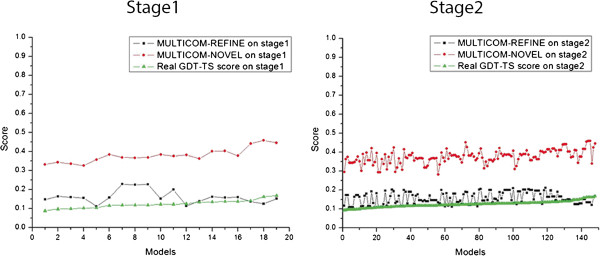
Results: MULTICOM-REFINE was a clustering approach that used the average pairwise structural similarity between models to measure the global quality and the average Euclidean distance between a model and several top ranked models to measure the local quality. MULTICOM-CLUSTER and MULTICOM-NOVEL were two new support vector machine-based methods of predicting both the local and global quality of a single protein model. MULTICOM-CONSTRUCT was a new weighted pairwise model comparison (clustering) method that used the weighted average similarity between models in a pool to measure the global model quality. Our experiments showed that the pairwise model assessment methods worked better when a large portion of models in the pool were of good quality, whereas single-model quality assessment methods performed better on some hard targets when only a small portion of models in the pool were of reasonable quality.
Conclusions: Since digging out a few good models from a large pool of low-quality models is a major challenge in protein structure prediction, single model quality assessment methods appear to be poised to make important contributions to protein structure modeling. The other interesting finding was that single-model quality assessment scores could be used to weight the models by the consensus pairwise model comparison method to improve its accuracy.
Figures
References
-
- Rost B. Protein structure prediction in 1D, 2D, and 3D. Encyclopaedia Comput Chem. 1998;3:2242–2255.
-
- Sali A, Blundell T. Comparative protein modelling by satisfaction of spatial restraints. Protein Structure Distance Anal. 1994;64:C86. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
