

Comparative genomic analysis and virulence differences in closely related salmonella enterica serotype heidelberg isolates from humans, retail meats, and animals
- PMID: 24732280
- PMCID: PMC4040988
- DOI: 10.1093/gbe/evu079
Comparative genomic analysis and virulence differences in closely related salmonella enterica serotype heidelberg isolates from humans, retail meats, and animals
Abstract
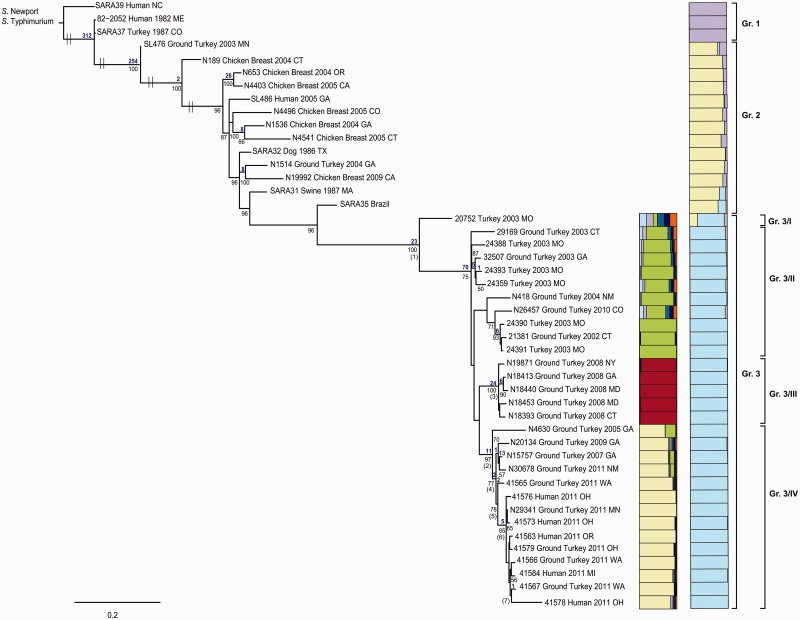
Salmonella enterica subsp. enterica serovar Heidelberg (S. Heidelberg) is one of the top serovars causing human salmonellosis. Recently, an antibiotic-resistant strain of this serovar was implicated in a large 2011 multistate outbreak resulting from consumption of contaminated ground turkey that involved 136 confirmed cases, with one death. In this study, we assessed the evolutionary diversity of 44 S. Heidelberg isolates using whole-genome sequencing (WGS) generated by the 454 GS FLX (Roche) platform. The isolates, including 30 with nearly indistinguishable (one band difference) Xbal pulsed-field gel electrophoresis patterns (JF6X01.0032, JF6X01.0058), were collected from various sources between 1982 and 2011 and included nine isolates associated with the 2011 outbreak. Additionally, we determined the complete sequence for the chromosome and three plasmids from a clinical isolate associated with the 2011 outbreak using the Pacific Biosciences (PacBio) system. Using single-nucleotide polymorphism (SNP) analyses, we were able to distinguish highly clonal isolates, including strains isolated at different times in the same year. The isolates from the recent 2011 outbreak clustered together with a mean SNP variation of only 17 SNPs. The S. Heidelberg isolates carried a variety of phages, such as prophage P22, P4, lambda-like prophage Gifsy-2, and the P2-like phage which carries the sopE1 gene, virulence genes including 62 pathogenicity, and 13 fimbrial markers and resistance plasmids of the incompatibility (Inc)I1, IncA/C, and IncHI2 groups. Twenty-one strains contained an IncX plasmid carrying a type IV secretion system. On the basis of the recent and historical isolates used in this study, our results demonstrated that, in addition to providing detailed genetic information for the isolates, WGS can identify SNP targets that can be utilized for differentiating highly clonal S. Heidelberg isolates.
Keywords: SNP analysis; antimicrobial resistance; outbreak; plasmid; trace-back.
Figures







References
-
- Aballay A, Yorgey P, Ausubel FM. Salmonella typhimurium proliferates and establishes a persistent infection in the intestine of Caenorhabditis elegans. Curr Biol. 2000;10:1539–1542. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
