

Extensive and coordinated control of allele-specific expression by both transcription and translation in Candida albicans
- PMID: 24732588
- PMCID: PMC4032860
- DOI: 10.1101/gr.166322.113
Extensive and coordinated control of allele-specific expression by both transcription and translation in Candida albicans
Abstract
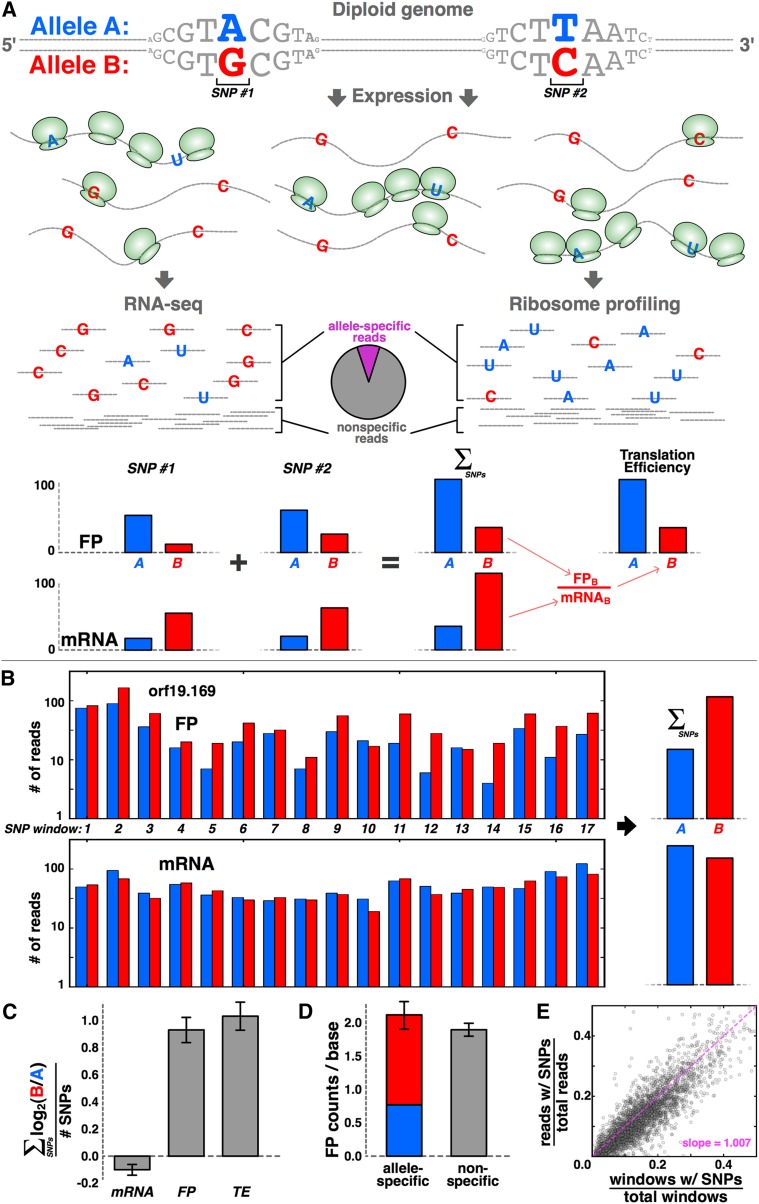
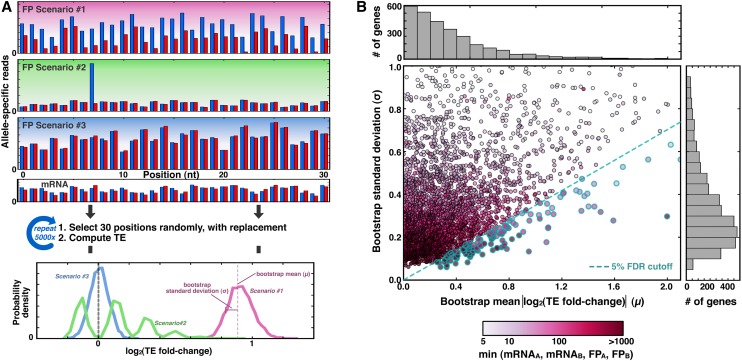
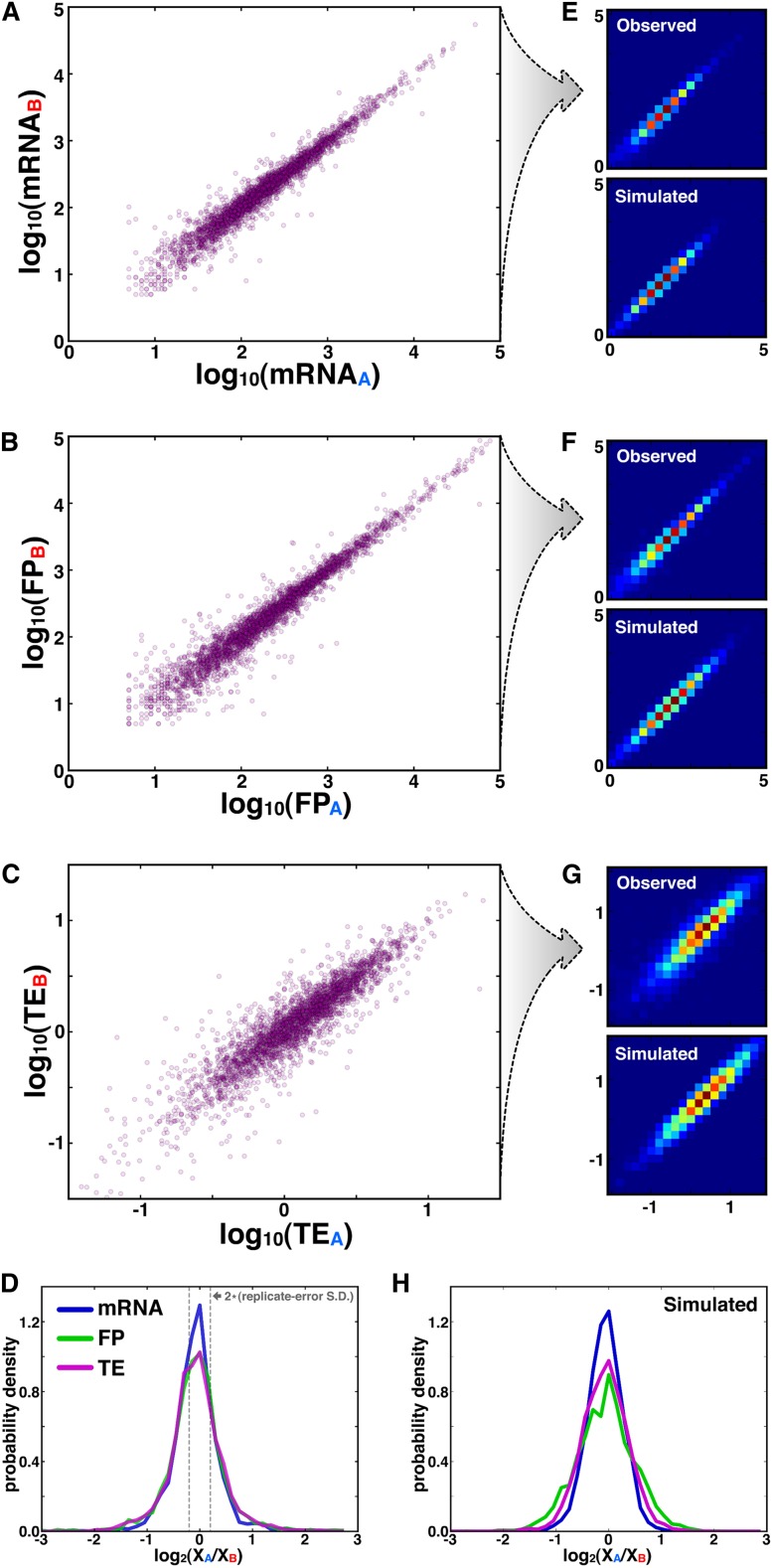
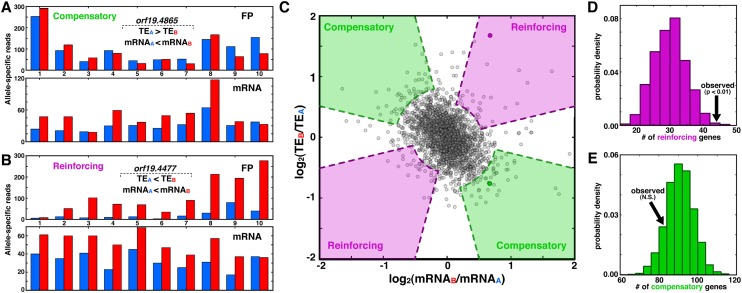
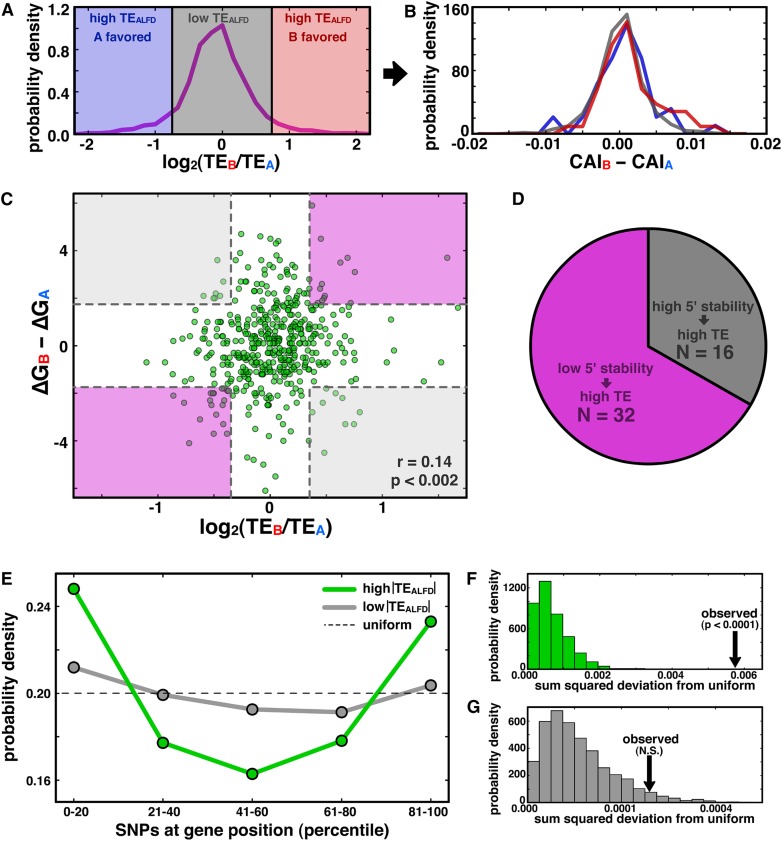
Though sequence differences between alleles are often limited to a few polymorphisms, these differences can cause large and widespread allelic variation at the expression level. Such allele-specific expression (ASE) has been extensively explored at the level of transcription but not translation. Here we measured ASE in the diploid yeast Candida albicans at both the transcriptional and translational levels using RNA-seq and ribosome profiling, respectively. Since C. albicans is an obligate diploid, our analysis isolates ASE arising from cis elements in a natural, nonhybrid organism, where allelic effects reflect evolutionary forces. Importantly, we find that ASE arising from translation is of a similar magnitude as transcriptional ASE, both in terms of the number of genes affected and the magnitude of the bias. We further observe coordination between ASE at the levels of transcription and translation for single genes. Specifically, reinforcing relationships--where transcription and translation favor the same allele--are more frequent than expected by chance, consistent with selective pressure tuning ASE at multiple regulatory steps. Finally, we parameterize alleles based on a range of properties and find that SNP location and predicted mRNA-structure stability are associated with translational ASE in cis. Since this analysis probes more than 4000 allelic pairs spanning a broad range of variations, our data provide a genome-wide view into the relative impact of cis elements that regulate translation.
© 2014 Muzzey et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Berriz GF, King OD, Bryant B, Sander C, Roth FP 2003. Characterizing gene sets with FuncAssociate. Bioinformatics 19: 2502–2504 - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases