

High glucose induces CCL20 in proximal tubular cells via activation of the KCa3.1 channel
- PMID: 24733189
- PMCID: PMC3986377
- DOI: 10.1371/journal.pone.0095173
High glucose induces CCL20 in proximal tubular cells via activation of the KCa3.1 channel
Abstract
Background: Inflammation plays a key role in the development and progression of diabetic nephropathy (DN). KCa3.1, a calcium activated potassium channel protein, is associated with vascular inflammation, atherogenesis, and proliferation of endothelial cells, macrophages, and fibroblasts. We have previously demonstrated that the KCa3.1 channel is activated by TGF-β1 and blockade of KCa3.1 ameliorates renal fibrotic responses in DN through inhibition of the TGF-β1 pathway. The present study aimed to identify the role of KCa3.1 in the inflammatory responses inherent in DN.
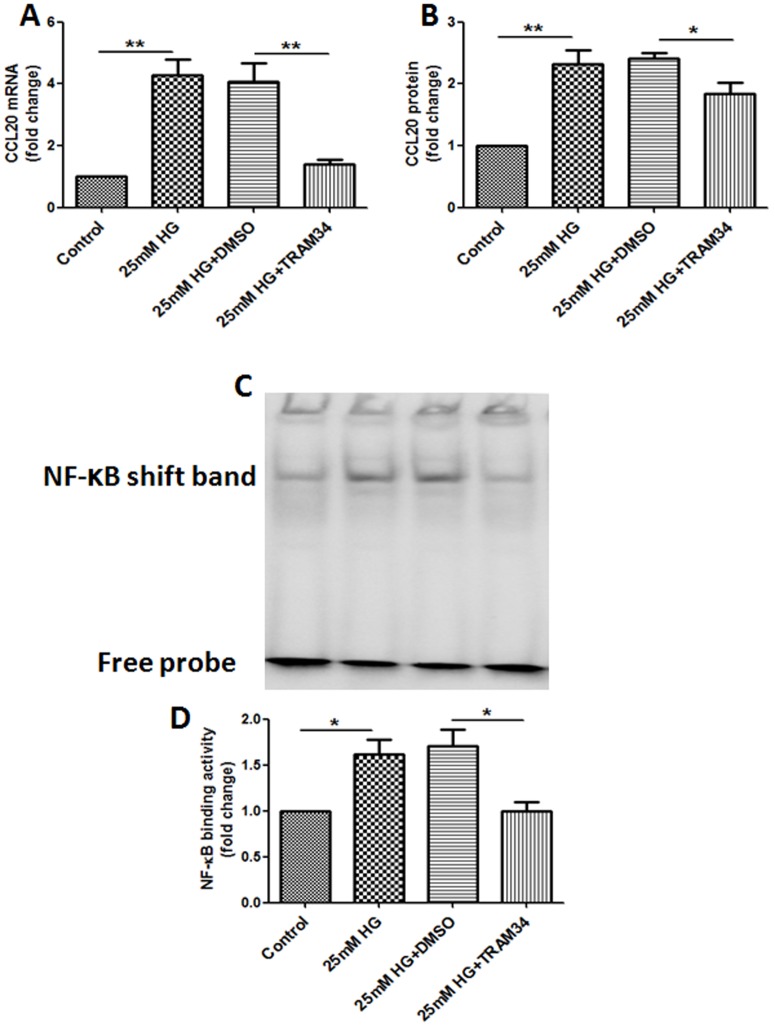
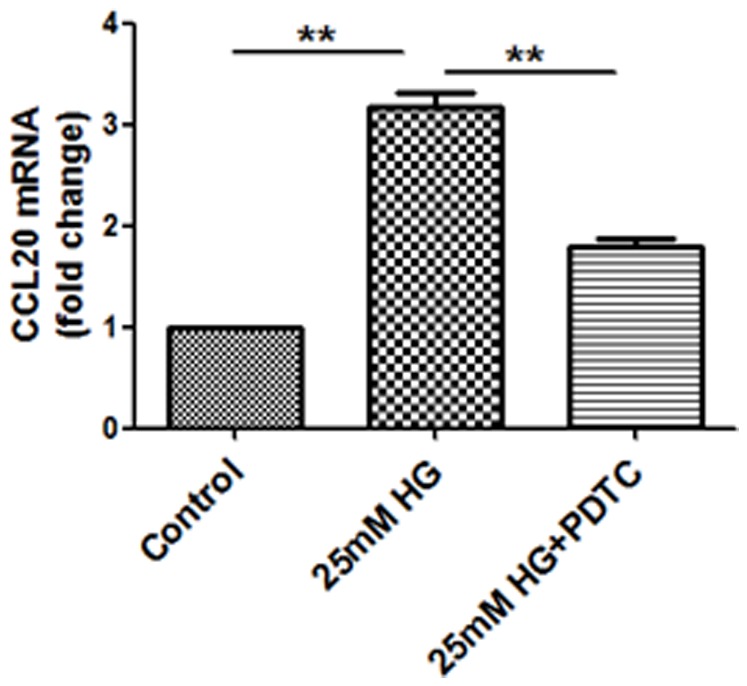
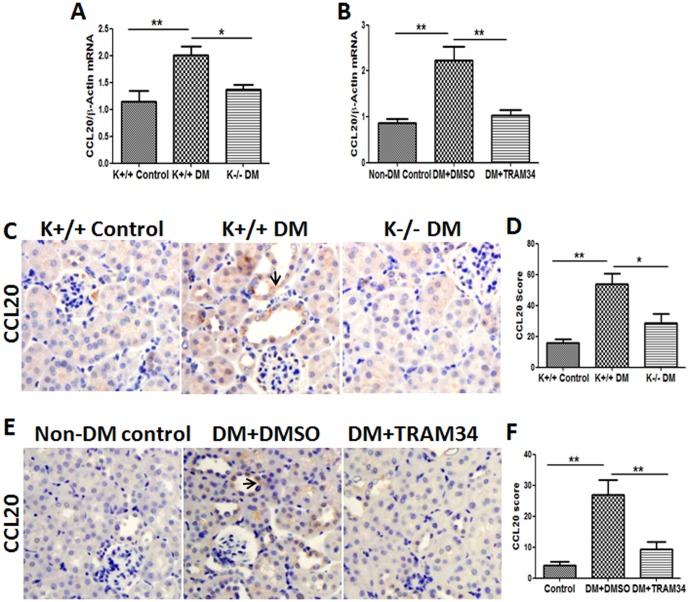
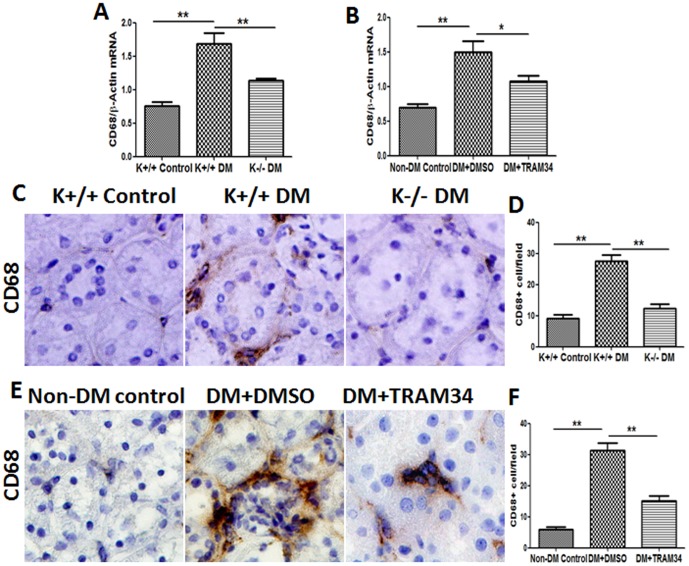
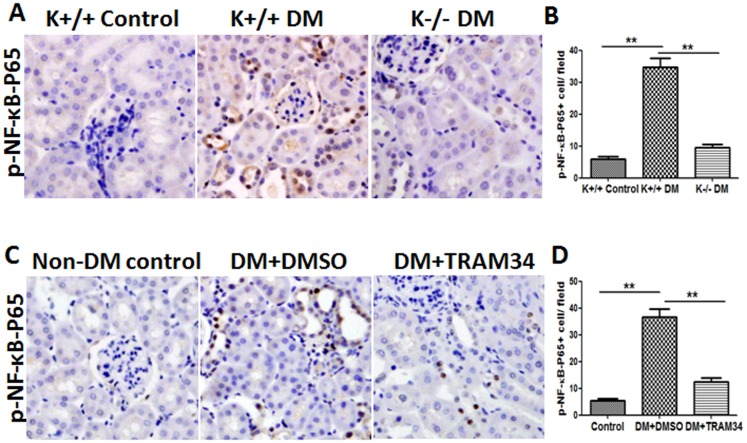
Methods: Human proximal tubular cells (HK2 cells) were exposed to high glucose (HG) in the presence or absence of the KCa3.1 inhibitor TRAM34 for 6 days. The proinflammatory cytokine chemokine (C-C motif) ligand 20 (CCL20) expression was examined by real-time PCR and enzyme-linked immunosorbent assay (ELISA). The activity of nuclear factor-κB (NF-κB) was measured by nuclear extraction and electrophoretic mobility shift assay (EMSA). In vivo, the expression of CCL20, the activity of NF-κB and macrophage infiltration (CD68 positive cells) were examined by real-time PCR and/or immunohistochemistry staining in kidneys from diabetic or KCa3.1-/- mice, and in eNOS-/- diabetic mice treated with the KCa3.1 channel inhibitor TRAM34.
Results: In vitro data showed that TRAM34 inhibited CCL20 expression and NF-κB activation induced by HG in HK2 cells. Both mRNA and protein levels of CCL20 significantly decreased in kidneys of diabetic KCa3.1-/- mice compared to diabetic wild type mice. Similarly, TRAM34 reduced CCL20 expression and NF-κB activation in diabetic eNOS-/- mice compared to diabetic controls. Blocking the KCa3.1 channel in both animal models led to a reduction in phosphorylated NF-κB.
Conclusions: Overexpression of CCL20 in human proximal tubular cells is inhibited by blockade of KCa3.1 under diabetic conditions through inhibition of the NF-κB pathway.
Conflict of interest statement
Figures
Similar articles
-
Inhibition of KCa3.1 suppresses TGF-β1 induced MCP-1 expression in human proximal tubular cells through Smad3, p38 and ERK1/2 signaling pathways.Int J Biochem Cell Biol. 2014 Feb;47:1-10. doi: 10.1016/j.biocel.2013.11.017. Epub 2013 Nov 28. Int J Biochem Cell Biol. 2014. PMID: 24291552
-
Blockade of KCa3.1 ameliorates renal fibrosis through the TGF-β1/Smad pathway in diabetic mice.Diabetes. 2013 Aug;62(8):2923-34. doi: 10.2337/db13-0135. Epub 2013 May 8. Diabetes. 2013. PMID: 23656889 Free PMC article.
-
KCa3.1 mediates activation of fibroblasts in diabetic renal interstitial fibrosis.Nephrol Dial Transplant. 2014 Feb;29(2):313-24. doi: 10.1093/ndt/gft431. Epub 2013 Oct 28. Nephrol Dial Transplant. 2014. PMID: 24166472 Free PMC article.
-
KCa3.1: a new player in progressive kidney disease.Curr Opin Nephrol Hypertens. 2015 Jan;24(1):61-6. doi: 10.1097/MNH.0000000000000083. Curr Opin Nephrol Hypertens. 2015. PMID: 25415613 Review.
-
Role of the potassium channel KCa3.1 in diabetic nephropathy.Clin Sci (Lond). 2014 Oct;127(7):423-33. doi: 10.1042/CS20140075. Clin Sci (Lond). 2014. PMID: 24963668 Review.
Cited by
-
The KCa3.1 blocker TRAM34 reverses renal damage in a mouse model of established diabetic nephropathy.PLoS One. 2018 Feb 9;13(2):e0192800. doi: 10.1371/journal.pone.0192800. eCollection 2018. PLoS One. 2018. PMID: 29425253 Free PMC article.
-
Tacrolimus ameliorates tubulointerstitial inflammation in diabetic nephropathy via inhibiting the NFATc1/TRPC6 pathway.J Cell Mol Med. 2020 Sep;24(17):9810-9824. doi: 10.1111/jcmm.15562. Epub 2020 Aug 11. J Cell Mol Med. 2020. PMID: 32779844 Free PMC article.
-
KCa3.1 mediates dysfunction of tubular autophagy in diabetic kidneys via PI3k/Akt/mTOR signaling pathways.Sci Rep. 2016 Mar 31;6:23884. doi: 10.1038/srep23884. Sci Rep. 2016. PMID: 27029904 Free PMC article.
-
Ion channels as a therapeutic target for renal fibrosis.Front Physiol. 2022 Oct 5;13:1019028. doi: 10.3389/fphys.2022.1019028. eCollection 2022. Front Physiol. 2022. PMID: 36277193 Free PMC article. Review.
-
Admission glucose, HbA1c levels and inflammatory cytokines in patients with acute ST-elevation myocardial infarction.Clin Proteomics. 2025 Feb 17;22(1):8. doi: 10.1186/s12014-025-09530-y. Clin Proteomics. 2025. PMID: 39962379 Free PMC article.
References
-
- Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C, et al. (2004) Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J Biol Chem 279: 47681–47687. - PubMed
-
- Chou CC, Lunn CA, Murgolo NJ (2008) KCa3.1: target and marker for cancer, autoimmune disorder and vascular inflammation? Expert Rev Mol Diagn 8: 179–187. - PubMed
-
- Hu L, Pennington M, Jiang Q, Whartenby KA, Calabresi PA (2007) Characterization of the functional properties of the voltage-gated potassium channel Kv1.3 in human CD4+ T lymphocytes. J Immunol 179: 4563–4570. - PubMed
-
- Tharp DL, Bowles DK (2009) The intermediate-conductance Ca2+ -activated K+ channel (KCa3.1) in vascular disease. Cardiovasc Hematol Agents Med Chem 7: 1–11. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
