

ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases
- PMID: 24735413
- PMCID: PMC4028082
- DOI: 10.1186/1471-2164-15-284
ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases
Abstract
Background: Understanding the relationship between the millions of functional DNA elements and their protein regulators, and how they work in conjunction to manifest diverse phenotypes, is key to advancing our understanding of the mammalian genome. Next-generation sequencing technology is now used widely to probe these protein-DNA interactions and to profile gene expression at a genome-wide scale. As the cost of DNA sequencing continues to fall, the interpretation of the ever increasing amount of data generated represents a considerable challenge.
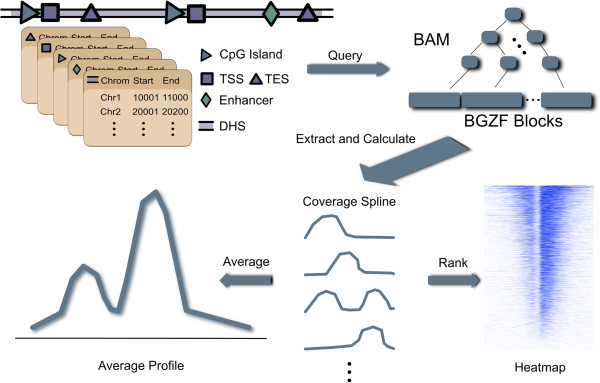
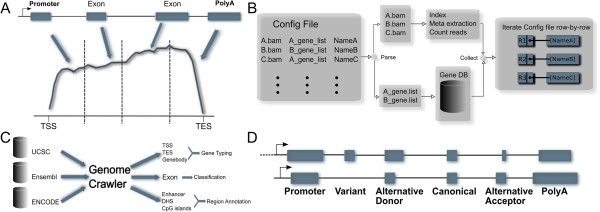
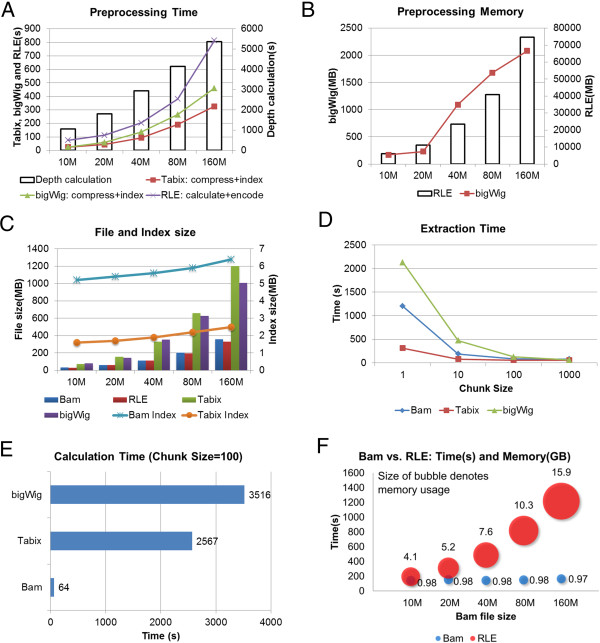
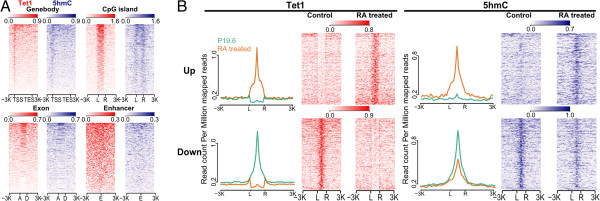
Results: We have developed ngs.plot - a standalone program to visualize enrichment patterns of DNA-interacting proteins at functionally important regions based on next-generation sequencing data. We demonstrate that ngs.plot is not only efficient but also scalable. We use a few examples to demonstrate that ngs.plot is easy to use and yet very powerful to generate figures that are publication ready.
Conclusions: We conclude that ngs.plot is a useful tool to help fill the gap between massive datasets and genomic information in this era of big sequencing data.
Figures
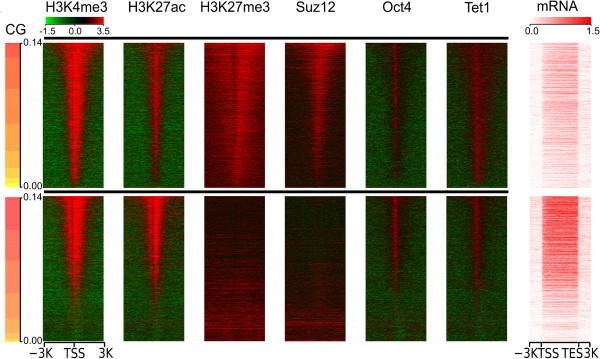
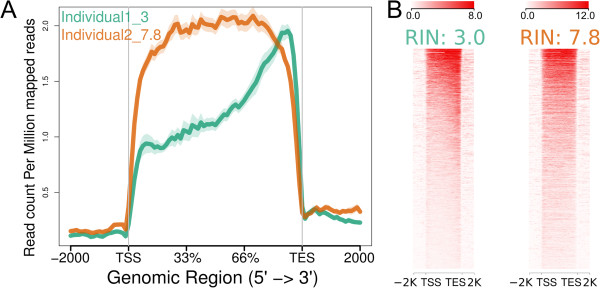
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
