

Clinical spectrum of females with HCCS mutation: from no clinical signs to a neonatal lethal form of the microphthalmia with linear skin defects (MLS) syndrome
- PMID: 24735900
- PMCID: PMC4021606
- DOI: 10.1186/1750-1172-9-53
Clinical spectrum of females with HCCS mutation: from no clinical signs to a neonatal lethal form of the microphthalmia with linear skin defects (MLS) syndrome
Abstract
Background: Segmental Xp22.2 monosomy or a heterozygous HCCS mutation is associated with the microphthalmia with linear skin defects (MLS) or MIDAS (microphthalmia, dermal aplasia, and sclerocornea) syndrome, an X-linked disorder with male lethality. HCCS encodes the holocytochrome c-type synthase involved in mitochondrial oxidative phosphorylation (OXPHOS) and programmed cell death.
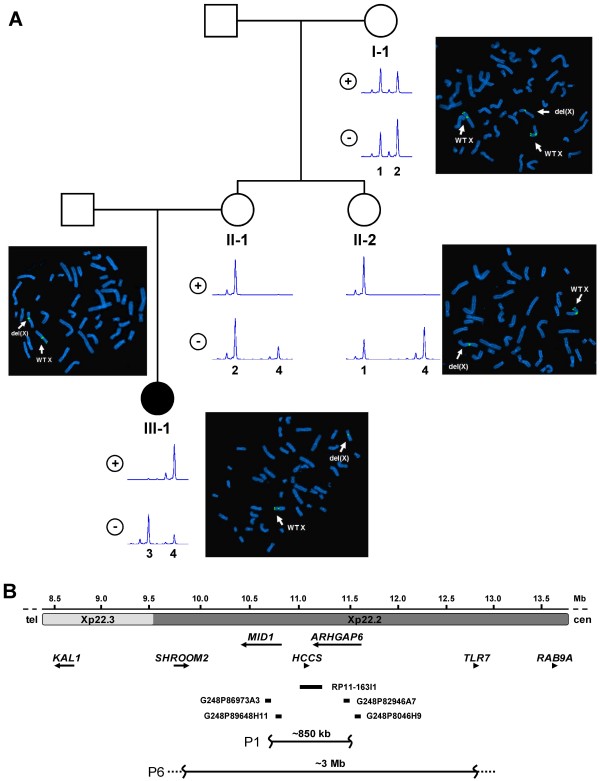
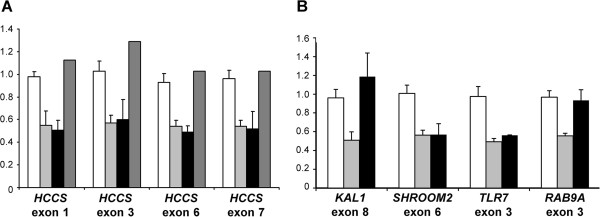
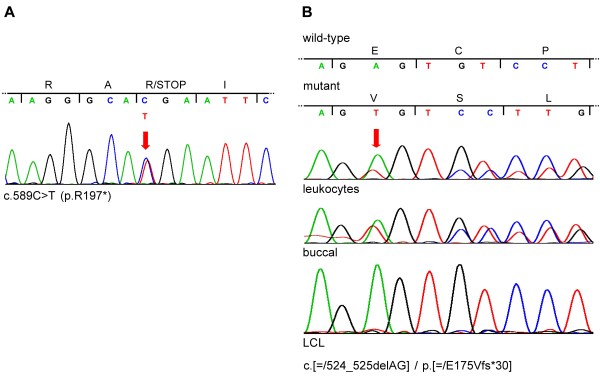
Methods: We characterized the X-chromosomal abnormality encompassing HCCS or an intragenic mutation in this gene in six new female patients with an MLS phenotype by cytogenetic analysis, fluorescence in situ hybridization, sequencing, and quantitative real-time PCR. The X chromosome inactivation (XCI) pattern was determined and clinical data of the patients were reviewed.
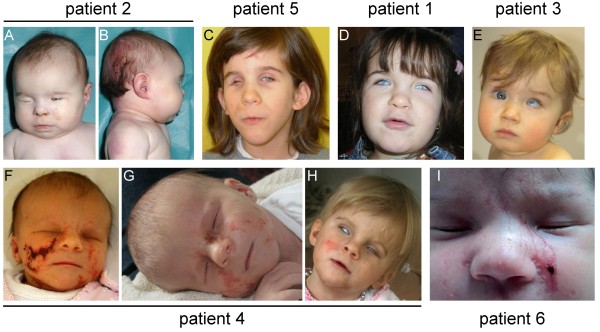
Results: Two terminal Xp deletions of ≥ 11.2 Mb, two submicroscopic copy number losses, one of ~850 kb and one of ≥ 3 Mb, all covering HCCS, 1 nonsense, and one mosaic 2-bp deletion in HCCS are reported. All females had a completely (>98:2) or slightly skewed (82:18) XCI pattern. The most consistent clinical features were microphthalmia/anophthalmia and sclerocornea/corneal opacity in all patients and congenital linear skin defects in 4/6. Additional manifestations included various ocular anomalies, cardiac defects, brain imaging abnormalities, microcephaly, postnatal growth retardation, and facial dysmorphism. However, no obvious clinical sign was observed in three female carriers who were relatives of one patient.
Conclusion: Our findings showed a wide phenotypic spectrum ranging from asymptomatic females with an HCCS mutation to patients with a neonatal lethal MLS form. Somatic mosaicism and the different ability of embryonic cells to cope with an OXPHOS defect and/or enhanced cell death upon HCCS deficiency likely underlie the great variability in phenotypes.
Figures
Similar articles
-
Mother and daughter with a terminal Xp deletion: implication of chromosomal mosaicism and X-inactivation in the high clinical variability of the microphthalmia with linear skin defects (MLS) syndrome.Eur J Med Genet. 2007 Nov-Dec;50(6):421-31. doi: 10.1016/j.ejmg.2007.07.004. Epub 2007 Aug 6. Eur J Med Genet. 2007. PMID: 17845869
-
Mutations in NDUFB11, encoding a complex I component of the mitochondrial respiratory chain, cause microphthalmia with linear skin defects syndrome.Am J Hum Genet. 2015 Apr 2;96(4):640-50. doi: 10.1016/j.ajhg.2015.02.002. Epub 2015 Mar 12. Am J Hum Genet. 2015. PMID: 25772934 Free PMC article.
-
Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome.Am J Hum Genet. 2006 Nov;79(5):878-89. doi: 10.1086/508474. Epub 2006 Sep 6. Am J Hum Genet. 2006. PMID: 17033964 Free PMC article.
-
Linear Skin Defects with Multiple Congenital Anomalies (LSDMCA): An Unconventional Mitochondrial Disorder.Genes (Basel). 2021 Feb 11;12(2):263. doi: 10.3390/genes12020263. Genes (Basel). 2021. PMID: 33670341 Free PMC article. Review.
-
Microphthalmia with linear skin defects syndrome in a mosaic female infant with monosomy for the Xp22 region: molecular analysis of the Xp22 breakpoint and the X-inactivation pattern.Hum Genet. 1998 Jul;103(1):51-6. doi: 10.1007/s004390050782. Hum Genet. 1998. PMID: 9737776 Review.
Cited by
-
Structural Insights into Mechanisms Underlying Mitochondrial and Bacterial Cytochrome c Synthases.Biomolecules. 2024 Nov 21;14(12):1483. doi: 10.3390/biom14121483. Biomolecules. 2024. PMID: 39766190 Free PMC article.
-
A mosaic form of microphthalmia with linear skin defects.BMC Pediatr. 2018 Aug 1;18(1):254. doi: 10.1186/s12887-018-1234-4. BMC Pediatr. 2018. PMID: 30068298 Free PMC article.
-
A Novel 4-Gene Score to Predict Survival, Distant Metastasis and Response to Neoadjuvant Therapy in Breast Cancer.Cancers (Basel). 2020 May 2;12(5):1148. doi: 10.3390/cancers12051148. Cancers (Basel). 2020. PMID: 32370309 Free PMC article.
-
Microphthalmia, Linear Skin Defects, Callosal Agenesis, and Cleft Palate in a Patient with Deletion at Xp22.3p22.2.J Pediatr Genet. 2020 Dec;9(4):258-262. doi: 10.1055/s-0039-3402047. Epub 2020 Jan 3. J Pediatr Genet. 2020. PMID: 32765930 Free PMC article.
-
Spectrum of combined respiratory chain defects.J Inherit Metab Dis. 2015 Jul;38(4):629-40. doi: 10.1007/s10545-015-9831-y. Epub 2015 Mar 17. J Inherit Metab Dis. 2015. PMID: 25778941 Free PMC article. Review.
References
-
- Morleo M, Franco B. In: GeneReviews™. Pagon RA AM, Bird TD, Dolan CR, Fong CT, Stephens K, editor. Seattle: University of Washington, Seattle; 2009. Microphthalmia with Linear Skin Defects Syndrome. - PubMed
-
- Wapenaar MC, Bassi MT, Schaefer L, Grillo A, Ferrero GB, Chinault AC, Ballabio A, Zoghbi HY. The genes for X-linked ocular albinism (OA1) and microphthalmia with linear skin defects (MLS): cloning and characterization of the critical regions. Hum Mol Genet. 1993;2:947–952. doi: 10.1093/hmg/2.7.947. - DOI - PubMed
-
- Wimplinger I, Morleo M, Rosenberger G, Iaconis D, Orth U, Meinecke P, Lerer I, Ballabio A, Gal A, Franco B, Kutsche K. Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome. Am J Hum Genet. 2006;79:878–889. doi: 10.1086/508474. - DOI - PMC - PubMed
-
- Wimplinger I, Shaw GM, Kutsche K. HCCS loss-of-function missense mutation in a female with bilateral microphthalmia and sclerocornea: a novel gene for severe ocular malformations? Mol Vis. 2007;13:1475–1482. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
