

Recent advances in host defense mechanisms/therapies against oral infectious diseases and consequences for systemic disease
- PMID: 24736702
- PMCID: PMC6636230
- DOI: 10.1177/0022034514525778
Recent advances in host defense mechanisms/therapies against oral infectious diseases and consequences for systemic disease
Abstract
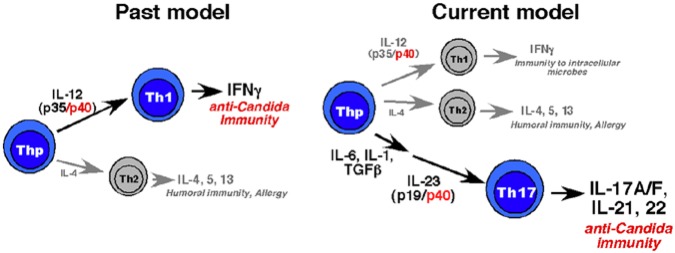
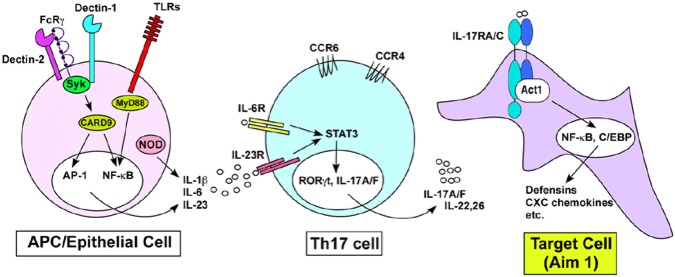
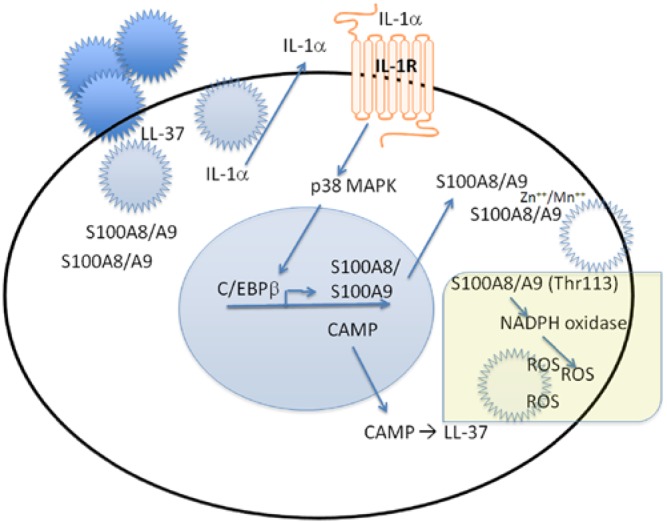
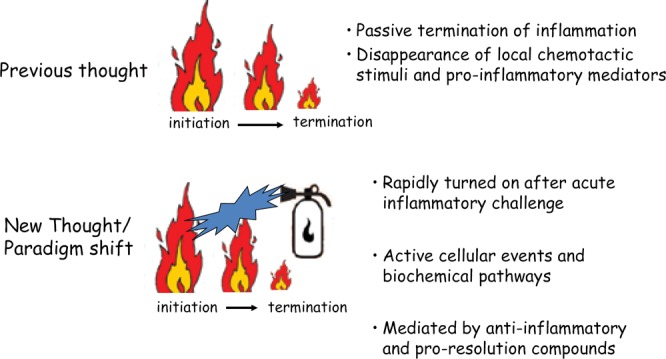
The innate and adaptive immune systems are both crucial to oral disease mechanisms and their impact on systemic health status. Greater understanding of these interrelationships will yield opportunities to identify new therapeutic targets to modulate disease processes and/or increase host resistance to infectious or inflammatory insult. The topics addressed reflect the latest advances in our knowledge of the role of innate and adaptive immune systems and inflammatory mechanisms in infectious diseases affecting the oral cavity, including periodontitis and candidiasis. In addition, several potential links with systemic inflammatory conditions, such as cardiovascular disease, are explored. The findings elucidate some of the defense mechanisms utilized by host tissues, including the role of IL-17 in providing immunity to oral candidiasis, the antimicrobial defense of mucosal epithelial cells, and the pro-resolution effects of the natural inflammatory regulators, proresolvins and lipoxins. They also describe the role of immune cells in mediating pathologic bone resorption in periodontal disease. These insights highlight the potential for therapeutic benefit of immunomodulatory interventions that bolster or modulate host defense mechanisms in both oral and systemic disease. Among the promising new therapeutic approaches discussed here are epithelial cell gene therapy, passive immunization against immune cell targets, and the use of proresolvin agents.
Keywords: bacterial invasion; cytokines; fungal immunity; lipoxins; oropharyngeal candidiasis; periodontal bone resorption.
Figures
References
-
- Asikainen P, Ruotsalainen TJ, Mikkonen JJ, Koistinen A, Ten Bruggenkate C, Kullaa AM. (2012). The defence architecture of the superficial cells of the oral mucosa. Med Hypotheses 78: 790–792. - PubMed
-
- Black RA. (2004). TIMP3 checks inflammation. Nat Genet 36: 934–935. - PubMed
-
- Brown GD. (2010). How fungi have shaped our understanding of mammalian immunology. Cell Host Microbe 7: 9–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 15566/PHS HHS/United States
- R01 DE022550/DE/NIDCR NIH HHS/United States
- R01DE021206/DE/NIDCR NIH HHS/United States
- R01 AI107825/AI/NIAID NIH HHS/United States
- DE19938/DE/NIDCR NIH HHS/United States
- R01 DE018499/DE/NIDCR NIH HHS/United States
- R01 DE023815/DE/NIDCR NIH HHS/United States
- DE018499/DE/NIDCR NIH HHS/United States
- R37 DE003420/DE/NIDCR NIH HHS/United States
- R01 DE015566/DE/NIDCR NIH HHS/United States
- DE021831/DE/NIDCR NIH HHS/United States
- R01 DE003420/DE/NIDCR NIH HHS/United States
- DE 03420/DE/NIDCR NIH HHS/United States
- R01 DE021206/DE/NIDCR NIH HHS/United States
- R01 DE019938/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
