

Distinct neurological disorders with ATP1A3 mutations
- PMID: 24739246
- PMCID: PMC4238309
- DOI: 10.1016/S1474-4422(14)70011-0
Distinct neurological disorders with ATP1A3 mutations
Abstract
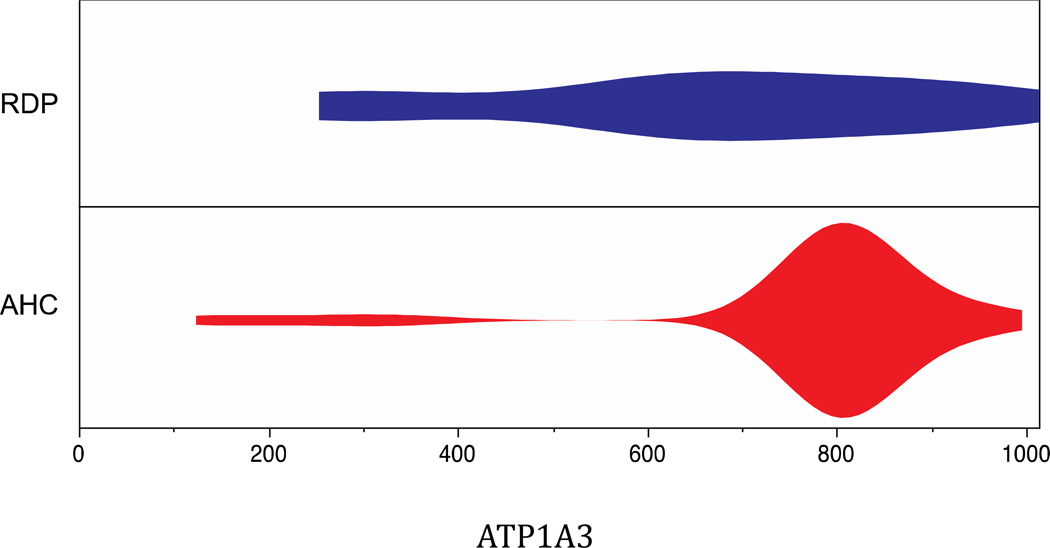
Genetic research has shown that mutations that modify the protein-coding sequence of ATP1A3, the gene encoding the α3 subunit of Na(+)/K(+)-ATPase, cause both rapid-onset dystonia parkinsonism and alternating hemiplegia of childhood. These discoveries link two clinically distinct neurological diseases to the same gene, however, ATP1A3 mutations are, with one exception, disease-specific. Although the exact mechanism of how these mutations lead to disease is still unknown, much knowledge has been gained about functional consequences of ATP1A3 mutations using a range of in-vitro and animal model systems, and the role of Na(+)/K(+)-ATPases in the brain. Researchers and clinicians are attempting to further characterise neurological manifestations associated with mutations in ATP1A3, and to build on the existing molecular knowledge to understand how specific mutations can lead to different diseases.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures




References
-
- Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14(10):681–691. - PubMed
-
- Bottger P, Tracz Z, Heuck A, Nissen P, Romero-Ramos M, Lykke-Hartmann K. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J Comp Neurol. 2011;519(2):376–404. - PubMed
-
- Dobretsov M, Stimers JR. Neuronal function and alpha3 isoform of the Na/K-ATPase. Frontiers in bioscience : a journal and virtual library. 2005;10:2373–2396. - PubMed
-
- Dobretsov M, Hastings SL, Sims TJ, Stimers JR, Romanovsky D. Stretch receptor-associated expression of alpha 3 isoform of the Na+, K+-ATPase in rat peripheral nervous system. Neuroscience. 2003;116(4):1069–1080. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
