

A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype
- PMID: 24739459
- PMCID: PMC4108029
- DOI: 10.1186/1471-2229-14-99
A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype
Abstract
Background: Alternative splicing (AS) significantly enhances transcriptome complexity. It is differentially regulated in a wide variety of cell types and plays a role in several cellular processes. Here we describe a detailed survey of alternative splicing in grape based on 124 SOLiD RNAseq analyses from different tissues, stress conditions and genotypes.
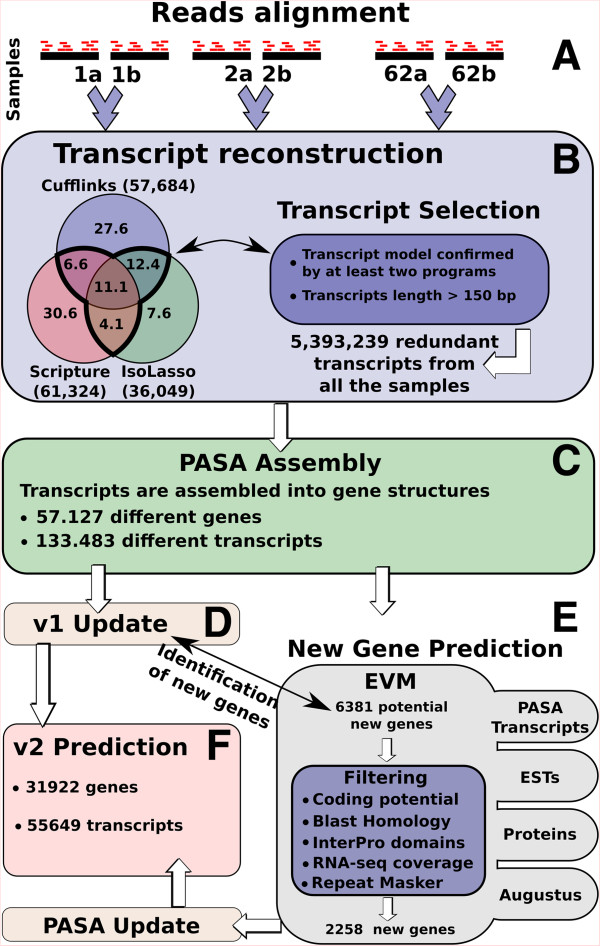
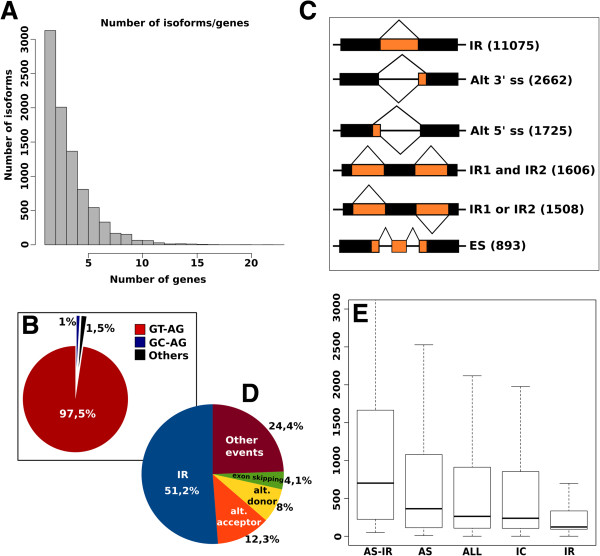
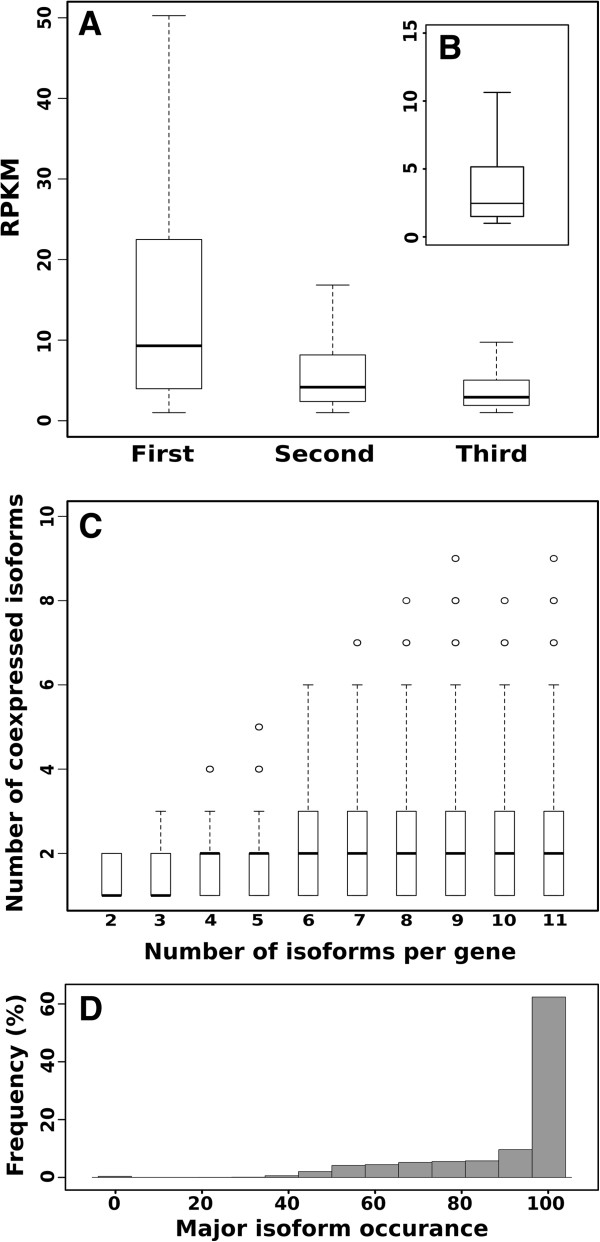
Results: We used the RNAseq data to update the existing grape gene prediction with 2,258 new coding genes and 3,336 putative long non-coding RNAs. Several gene structures have been improved and alternative splicing was described for about 30% of the genes. A link between AS and miRNAs was shown in 139 genes where we found that AS affects the miRNA target site. A quantitative analysis of the isoforms indicated that most of the spliced genes have one major isoform and tend to simultaneously co-express a low number of isoforms, typically two, with intron retention being the most frequent alternative splicing event.
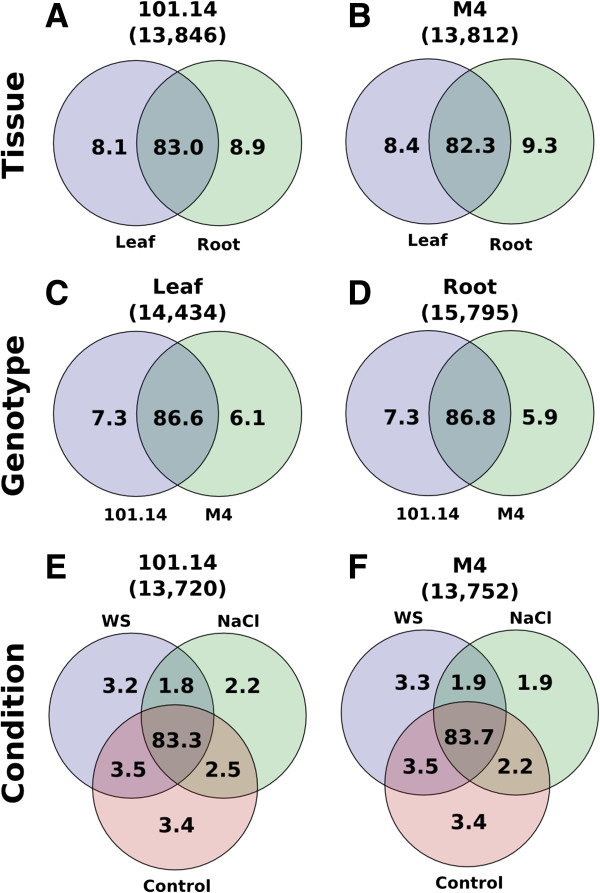
Conclusions: As described in Arabidopsis, also grape displays a marked AS tissue-specificity, while stress conditions produce splicing changes to a minor extent. Surprisingly, some distinctive splicing features were also observed between genotypes. This was further supported by the observation that the panel of Serine/Arginine-rich splicing factors show a few, but very marked differences between genotypes. The finding that a part the splicing machinery can change in closely related organisms can lead to some interesting hypotheses for evolutionary adaptation, that could be particularly relevant in the response to sudden and strong selective pressures.
Figures
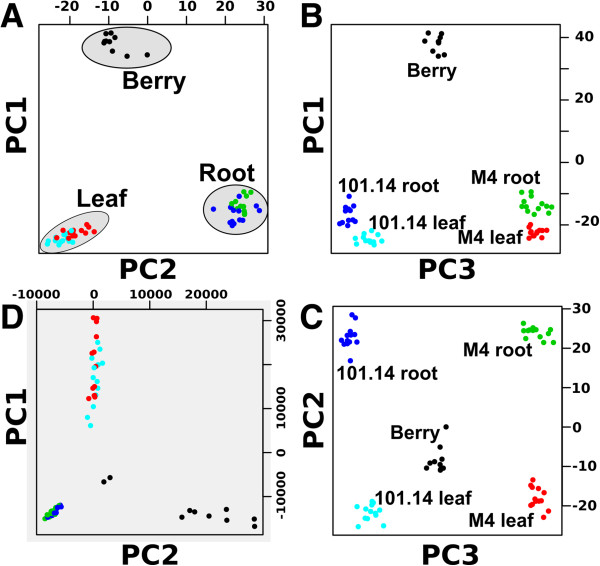
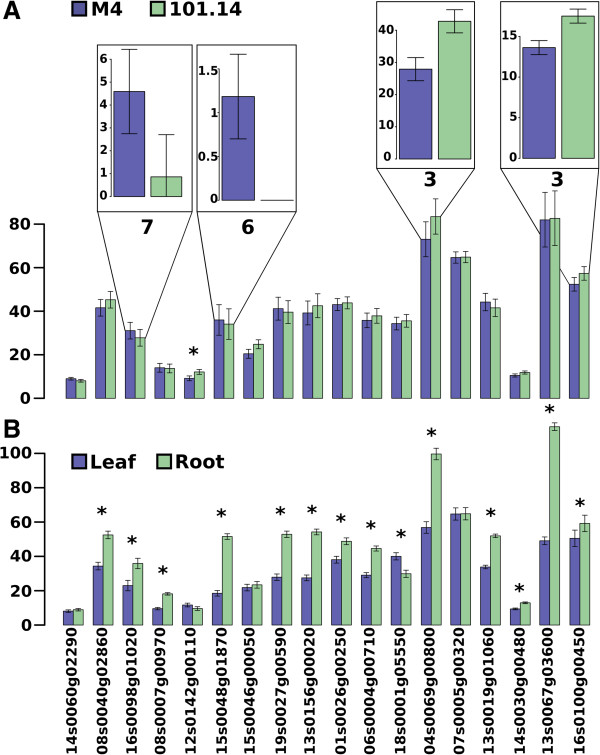
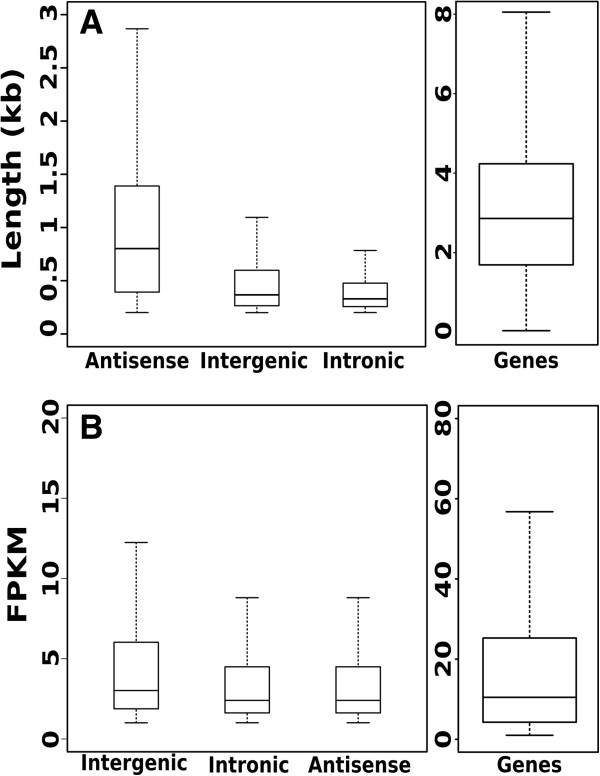
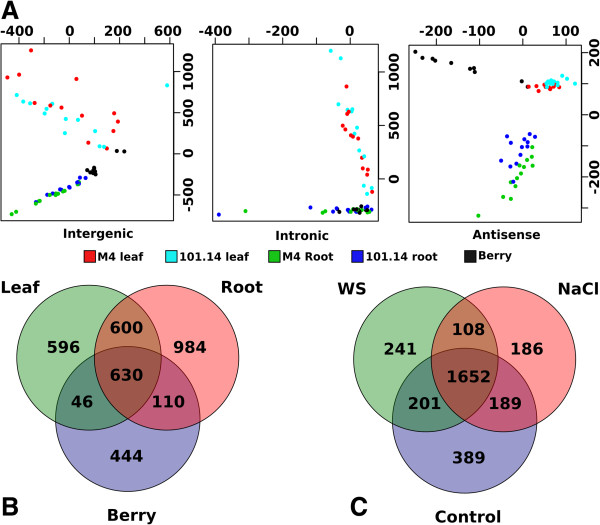
References
-
- Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V. et al.The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. - DOI - PubMed
-
- Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, Fitzgerald LM, Vezzulli S, Reid J, Malacarne G, Iliev D, Coppola G, Wardell B, Micheletti D, Macalma T, Facci M, Mitchell JT, Perazzolli M, Eldredge G, Gatto P, Oyzerski R, Moretto M, Gutin N, Stefanini M, Chen Y, Segala C, Davenport C, Demattè L, Mraz A. et al.A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One. 2007;2:e1326. doi: 10.1371/journal.pone.0001326. - DOI - PMC - PubMed
-
- Fasoli M, Dal Santo S, Zenoni S, Tornielli GB, Farina L, Zamboni A, Porceddu A, Venturini L, Bicego M, Murino V, Ferrarini A, Delledonne M, Pezzotti M. The grapevine expression atlas reveals a deep transcriptome shift driving the entire plant into a maturation program. Plant Cell. 2012;24:3489–3505. doi: 10.1105/tpc.112.100230. - DOI - PMC - PubMed
-
- Venturini L, Ferrarini A, Zenoni S, Tornielli GB, Fasoli M, Santo SD, Minio A, Buson G, Tononi P, Zago ED, Zamperin G, Bellin D, Pezzotti M, Delledonne M. De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity. BMC Genomics. 2013;14:41. doi: 10.1186/1471-2164-14-41. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
